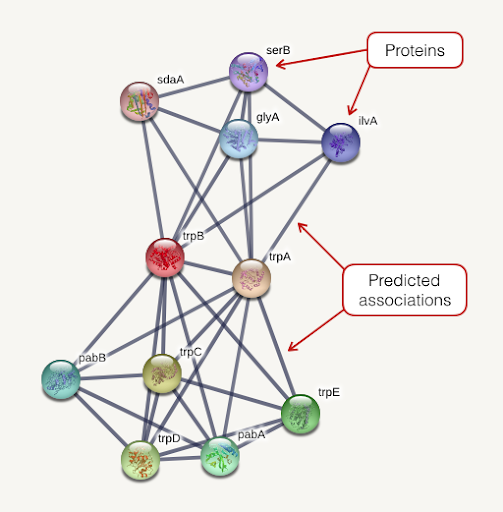
class: center, middle, inverse, title-slide .title[ # Lecture 9: Gene set enrichments ] .subtitle[ ## BE_22 Bioinformatics SS 21 ] .author[ ### January Weiner ] .date[ ### 2024-06-18 ] --- <!-- class:empty-slide,myinverse background-image:url(images/arnolfini.jpg) --> ## Gene set enrichment analysis The problem: how do you interpret a list of 1000 results? --- <div class="datatables html-widget html-fill-item-overflow-hidden html-fill-item" id="htmlwidget-55cfd73bed7608d44a24" style="width:100%;height:auto;"></div> <script type="application/json" data-for="htmlwidget-55cfd73bed7608d44a24">{"x":{"filter":"none","vertical":false,"data":[["1","2","3","4","5","6","7","8","9","10","11","12","13","14","15","16","17","18","19","20","21","22","23","24","25","26","27","28","29","30","31","32","33","34","35","36","37","38","39","40","41","42","43","44","45","46","47","48","49","50","51","52","53","54","55","56","57","58","59","60","61","62","63","64","65","66","67","68","69","70","71","72","73","74","75","76","77","78","79","80","81","82","83","84","85","86","87","88","89","90","91","92","93","94","95","96","97","98","99","100","101","102","103","104","105","106","107","108","109","110","111","112","113","114","115","116","117","118","119","120","121","122","123","124","125","126","127","128","129","130","131","132","133","134","135","136","137","138","139","140","141","142","143","144","145","146","147","148","149","150","151","152","153","154","155","156","157","158","159","160","161","162","163","164","165","166","167","168","169","170","171","172","173","174","175","176","177","178","179","180","181","182","183","184","185","186","187","188","189","190","191","192","193","194","195","196","197","198","199","200","201","202","203","204","205","206","207","208","209","210","211","212","213","214","215","216","217","218","219","220","221","222","223","224","225","226","227","228","229","230","231","232","233","234","235","236","237","238","239","240","241","242","243","244","245","246","247","248","249","250","251","252","253","254","255","256","257","258","259","260","261","262","263","264","265","266","267","268","269","270","271","272","273","274","275","276","277","278","279","280","281","282","283","284","285","286","287","288","289","290","291","292","293","294","295","296","297","298","299","300","301","302","303","304","305","306","307","308","309","310","311","312","313","314","315","316","317","318","319","320","321","322","323","324","325","326","327","328","329","330","331","332","333","334","335","336","337","338","339","340","341","342","343","344","345","346","347","348","349","350","351","352","353","354","355","356","357","358","359","360","361","362","363","364","365","366","367","368","369","370","371","372","373","374","375","376","377","378","379","380","381","382","383","384","385","386","387","388","389","390","391","392","393","394","395","396","397","398","399","400","401","402","403","404","405","406","407","408","409","410","411","412","413","414","415","416","417","418","419","420","421","422","423","424","425","426","427","428","429","430","431","432","433","434","435","436","437","438","439","440","441","442","443","444","445","446","447","448","449","450","451","452","453","454","455","456","457","458","459","460","461","462","463","464","465","466","467","468","469","470","471","472","473","474","475","476","477","478","479","480","481","482","483","484","485","486","487","488","489","490","491","492","493","494","495","496","497","498","499","500","501","502","503","504","505","506","507","508","509","510","511","512","513","514","515","516","517","518","519","520","521","522","523","524","525","526","527","528","529","530","531","532","533","534","535","536","537","538","539","540","541","542","543","544","545","546","547","548","549","550","551","552","553","554","555","556","557","558","559","560","561","562","563","564","565","566","567","568","569","570","571","572","573","574","575","576","577","578","579","580","581","582","583","584","585","586","587","588","589","590","591","592","593","594","595","596","597","598","599","600","601","602","603","604","605","606","607","608","609","610","611","612","613","614","615","616","617","618","619","620","621","622","623","624","625","626","627","628","629","630","631","632","633","634","635","636","637","638","639","640","641","642","643","644","645","646","647","648","649","650","651","652","653","654","655","656","657","658","659","660","661","662","663","664","665","666","667","668","669","670","671","672","673","674","675","676","677","678","679","680","681","682","683","684","685","686","687","688","689","690","691","692","693","694","695","696","697","698","699","700","701","702","703","704","705","706","707","708","709","710","711","712","713","714","715","716","717","718","719","720","721","722","723","724","725","726","727","728","729","730","731","732","733","734","735","736","737","738","739","740","741","742","743","744","745","746","747","748","749","750","751","752","753","754","755","756","757","758","759","760","761","762","763","764","765","766","767","768","769","770","771","772","773","774","775","776","777","778","779","780","781","782","783","784","785","786","787","788","789","790","791","792","793","794","795","796","797","798","799","800","801","802","803","804","805","806","807","808","809","810","811","812","813","814","815","816","817","818","819","820","821","822","823","824","825","826","827","828","829","830","831","832","833","834","835","836","837","838","839","840","841","842","843","844","845","846","847","848","849","850","851","852","853","854","855","856","857","858","859","860","861","862","863","864","865","866","867","868","869","870","871","872","873","874","875","876","877","878","879","880","881","882","883","884","885","886","887","888","889","890","891","892","893","894","895","896","897","898","899","900","901","902","903","904","905","906","907","908","909","910","911","912","913","914","915","916","917","918","919","920","921","922","923","924","925","926","927","928","929","930","931","932","933","934","935","936","937","938","939","940","941","942","943","944","945","946","947","948","949","950","951","952","953","954","955","956","957","958","959","960","961","962","963","964","965","966","967","968","969","970","971","972","973","974","975","976","977","978","979","980","981","982","983","984","985","986","987","988","989","990","991","992","993","994","995","996","997","998","999","1000","1001","1002","1003","1004","1005","1006","1007","1008","1009","1010","1011","1012","1013","1014","1015","1016","1017","1018","1019","1020","1021","1022","1023","1024","1025","1026","1027","1028","1029","1030","1031","1032","1033","1034","1035","1036","1037","1038","1039","1040","1041","1042","1043","1044","1045","1046","1047","1048","1049","1050","1051","1052","1053","1054","1055","1056","1057","1058","1059","1060","1061","1062","1063","1064","1065","1066","1067","1068","1069","1070","1071","1072","1073","1074","1075","1076","1077","1078","1079","1080","1081","1082","1083","1084","1085","1086","1087","1088","1089","1090","1091","1092","1093","1094","1095","1096","1097","1098","1099","1100","1101","1102","1103","1104","1105","1106","1107","1108","1109","1110","1111","1112","1113","1114","1115","1116","1117","1118","1119","1120","1121","1122","1123","1124","1125","1126","1127","1128","1129","1130","1131","1132","1133","1134","1135","1136","1137","1138","1139","1140","1141","1142","1143","1144","1145","1146","1147","1148","1149","1150","1151","1152","1153","1154","1155","1156","1157","1158","1159","1160","1161","1162","1163","1164","1165","1166","1167","1168","1169","1170","1171","1172","1173","1174","1175","1176","1177","1178","1179","1180","1181","1182","1183","1184","1185","1186","1187","1188","1189","1190","1191","1192","1193","1194","1195","1196","1197","1198","1199","1200","1201","1202","1203","1204","1205","1206","1207","1208","1209","1210","1211","1212","1213","1214","1215","1216","1217","1218","1219","1220","1221","1222","1223","1224","1225","1226","1227","1228","1229","1230","1231","1232","1233","1234","1235","1236","1237","1238","1239","1240","1241","1242","1243","1244","1245","1246","1247","1248","1249","1250","1251","1252","1253","1254","1255","1256","1257","1258","1259","1260","1261","1262","1263","1264","1265","1266","1267","1268","1269","1270","1271","1272","1273","1274","1275","1276","1277","1278","1279","1280","1281","1282","1283","1284","1285","1286","1287","1288","1289","1290","1291","1292","1293","1294","1295","1296","1297","1298","1299","1300","1301","1302","1303","1304","1305","1306","1307","1308","1309","1310","1311","1312","1313","1314","1315","1316","1317","1318","1319","1320","1321","1322","1323","1324","1325","1326","1327","1328","1329","1330","1331","1332","1333","1334","1335","1336","1337","1338","1339","1340","1341","1342","1343","1344","1345","1346","1347","1348","1349","1350","1351","1352","1353","1354","1355","1356","1357","1358","1359","1360","1361","1362","1363","1364","1365","1366","1367","1368","1369","1370","1371","1372","1373","1374","1375","1376","1377","1378","1379","1380","1381","1382","1383","1384","1385","1386","1387","1388","1389","1390","1391","1392","1393","1394","1395","1396","1397","1398","1399","1400","1401","1402","1403","1404","1405","1406","1407","1408","1409","1410","1411","1412","1413","1414","1415","1416","1417","1418","1419","1420","1421","1422","1423","1424","1425","1426","1427","1428","1429","1430","1431","1432","1433","1434","1435","1436","1437","1438","1439","1440","1441","1442","1443","1444","1445","1446","1447","1448","1449","1450","1451","1452","1453","1454","1455","1456","1457","1458","1459","1460","1461","1462","1463","1464","1465","1466","1467","1468","1469","1470","1471","1472","1473","1474","1475","1476","1477","1478","1479","1480","1481","1482","1483","1484","1485","1486","1487","1488","1489","1490","1491","1492","1493","1494","1495","1496","1497","1498","1499","1500","1501","1502","1503","1504","1505","1506","1507","1508","1509","1510","1511","1512","1513","1514","1515","1516","1517","1518","1519","1520","1521","1522","1523","1524","1525","1526","1527","1528","1529","1530","1531","1532","1533","1534","1535","1536","1537","1538","1539","1540","1541","1542","1543","1544","1545","1546","1547","1548","1549","1550","1551","1552","1553","1554","1555","1556","1557","1558","1559","1560","1561","1562","1563","1564","1565","1566","1567","1568","1569","1570","1571","1572","1573","1574","1575","1576","1577","1578","1579","1580","1581","1582","1583","1584","1585","1586","1587","1588","1589","1590","1591","1592","1593","1594","1595","1596","1597","1598","1599","1600","1601","1602","1603","1604","1605","1606","1607","1608","1609","1610","1611","1612","1613","1614","1615","1616","1617","1618","1619","1620","1621","1622","1623","1624","1625","1626","1627","1628","1629","1630","1631","1632","1633","1634","1635","1636","1637","1638","1639","1640","1641","1642","1643","1644","1645","1646","1647","1648","1649","1650","1651","1652","1653","1654","1655","1656","1657","1658","1659","1660","1661","1662","1663","1664","1665","1666","1667","1668","1669","1670","1671","1672","1673","1674","1675","1676","1677","1678","1679","1680","1681","1682","1683","1684","1685","1686","1687","1688","1689","1690","1691","1692","1693","1694","1695","1696","1697","1698","1699","1700","1701","1702","1703","1704","1705","1706","1707","1708","1709","1710","1711","1712","1713","1714","1715","1716","1717","1718","1719","1720","1721","1722","1723","1724","1725","1726","1727","1728","1729","1730","1731","1732","1733","1734","1735","1736","1737","1738","1739","1740","1741","1742","1743","1744","1745","1746","1747","1748","1749","1750","1751","1752","1753","1754","1755","1756","1757","1758","1759","1760","1761","1762","1763","1764","1765","1766","1767","1768","1769","1770","1771","1772","1773","1774","1775","1776","1777","1778","1779","1780","1781","1782","1783","1784","1785","1786","1787","1788","1789","1790","1791","1792","1793","1794","1795","1796","1797","1798","1799","1800","1801","1802","1803","1804","1805","1806","1807","1808","1809","1810","1811","1812","1813","1814","1815","1816","1817","1818","1819","1820","1821","1822","1823","1824","1825","1826","1827","1828","1829","1830","1831","1832","1833","1834","1835","1836","1837","1838","1839","1840","1841","1842","1843","1844","1845","1846","1847","1848","1849","1850","1851","1852","1853","1854","1855","1856","1857","1858","1859","1860","1861","1862","1863","1864","1865","1866","1867","1868","1869","1870","1871","1872","1873","1874","1875","1876","1877","1878","1879","1880","1881","1882","1883","1884","1885","1886","1887","1888","1889","1890","1891","1892","1893","1894","1895","1896","1897","1898","1899","1900","1901","1902","1903","1904","1905","1906","1907","1908","1909","1910","1911","1912","1913","1914","1915","1916","1917","1918","1919","1920","1921","1922","1923","1924","1925","1926","1927","1928","1929","1930","1931","1932","1933","1934","1935","1936","1937","1938","1939","1940","1941","1942","1943","1944","1945","1946","1947","1948","1949","1950","1951","1952","1953","1954","1955","1956","1957","1958","1959","1960","1961","1962","1963","1964","1965","1966","1967","1968","1969","1970","1971","1972","1973","1974","1975","1976","1977","1978","1979","1980","1981","1982","1983","1984","1985","1986","1987","1988","1989","1990","1991","1992","1993","1994","1995","1996","1997","1998","1999","2000","2001","2002","2003","2004","2005","2006","2007","2008","2009","2010","2011","2012","2013","2014","2015","2016","2017","2018","2019","2020","2021","2022","2023","2024","2025","2026","2027","2028","2029","2030","2031","2032","2033","2034","2035","2036","2037","2038","2039","2040","2041","2042","2043","2044","2045","2046","2047","2048","2049","2050","2051","2052","2053","2054","2055","2056","2057","2058","2059","2060","2061","2062","2063","2064","2065","2066","2067","2068","2069","2070","2071","2072","2073","2074","2075","2076","2077","2078","2079","2080","2081","2082","2083","2084","2085","2086","2087","2088","2089","2090","2091","2092","2093","2094","2095","2096","2097","2098","2099","2100","2101","2102","2103","2104","2105","2106","2107","2108","2109","2110","2111","2112","2113","2114","2115","2116","2117","2118","2119","2120","2121","2122","2123","2124","2125","2126","2127","2128","2129","2130","2131","2132","2133","2134","2135","2136","2137","2138","2139","2140","2141","2142","2143","2144","2145","2146","2147","2148","2149","2150","2151","2152","2153","2154","2155","2156","2157","2158","2159","2160","2161","2162","2163","2164","2165","2166","2167","2168","2169","2170","2171","2172","2173","2174","2175","2176","2177","2178","2179","2180","2181","2182","2183","2184","2185","2186","2187","2188","2189","2190","2191","2192","2193","2194","2195","2196","2197","2198","2199","2200","2201","2202","2203","2204","2205","2206","2207","2208","2209","2210","2211","2212","2213","2214","2215","2216","2217","2218","2219","2220","2221","2222","2223","2224","2225","2226","2227","2228","2229","2230","2231","2232","2233","2234","2235","2236","2237","2238","2239","2240","2241","2242","2243","2244","2245","2246","2247","2248","2249","2250","2251","2252","2253","2254","2255","2256","2257","2258","2259","2260","2261","2262","2263","2264","2265","2266","2267","2268","2269","2270","2271","2272","2273","2274","2275","2276","2277","2278","2279","2280","2281","2282","2283","2284","2285","2286","2287","2288","2289","2290","2291","2292","2293","2294","2295","2296","2297","2298","2299","2300","2301","2302","2303","2304","2305","2306","2307","2308","2309","2310","2311","2312","2313","2314","2315","2316","2317","2318","2319","2320","2321","2322","2323","2324","2325","2326","2327","2328","2329","2330","2331","2332","2333","2334","2335","2336","2337","2338","2339","2340","2341","2342","2343","2344","2345","2346","2347","2348","2349","2350","2351","2352","2353","2354","2355","2356","2357","2358","2359","2360","2361","2362","2363","2364","2365","2366","2367","2368","2369","2370","2371","2372","2373","2374","2375","2376","2377","2378","2379","2380","2381","2382","2383","2384","2385","2386","2387","2388","2389","2390","2391","2392","2393","2394","2395","2396","2397","2398","2399","2400","2401","2402","2403","2404","2405","2406","2407","2408","2409","2410","2411","2412","2413","2414","2415","2416","2417","2418","2419","2420","2421","2422","2423","2424","2425","2426","2427","2428","2429","2430","2431","2432","2433","2434","2435","2436","2437","2438","2439","2440","2441","2442","2443","2444","2445","2446","2447","2448","2449","2450","2451","2452","2453","2454","2455","2456","2457","2458","2459","2460","2461","2462","2463","2464","2465","2466","2467","2468","2469","2470","2471","2472","2473","2474","2475","2476","2477","2478","2479","2480","2481","2482","2483","2484","2485","2486","2487","2488","2489","2490","2491","2492","2493","2494","2495","2496","2497","2498","2499","2500","2501","2502","2503","2504","2505","2506","2507","2508","2509","2510","2511","2512","2513","2514","2515","2516","2517","2518","2519","2520","2521","2522","2523","2524","2525","2526","2527","2528","2529","2530","2531","2532","2533","2534","2535","2536","2537","2538","2539","2540","2541","2542","2543","2544","2545","2546","2547","2548","2549","2550","2551","2552","2553","2554","2555","2556","2557","2558","2559","2560","2561","2562","2563","2564","2565","2566","2567","2568","2569","2570","2571","2572","2573","2574","2575","2576","2577","2578","2579","2580","2581","2582","2583","2584","2585","2586","2587","2588","2589","2590","2591","2592","2593","2594","2595","2596","2597","2598","2599","2600","2601","2602","2603","2604","2605","2606","2607","2608","2609","2610","2611","2612","2613","2614","2615","2616","2617","2618","2619","2620","2621","2622","2623","2624","2625","2626","2627","2628","2629","2630","2631","2632","2633","2634","2635","2636","2637","2638","2639","2640","2641","2642","2643","2644","2645","2646","2647","2648","2649","2650","2651","2652","2653","2654","2655","2656","2657","2658","2659","2660","2661","2662","2663","2664","2665","2666","2667","2668","2669","2670","2671","2672","2673","2674","2675","2676","2677","2678","2679","2680","2681","2682","2683","2684","2685","2686","2687","2688","2689","2690","2691","2692","2693","2694","2695","2696","2697","2698","2699","2700","2701","2702","2703","2704","2705","2706","2707","2708","2709","2710","2711","2712","2713","2714","2715","2716","2717","2718","2719","2720","2721","2722","2723","2724","2725","2726","2727","2728","2729","2730","2731","2732","2733","2734","2735","2736","2737","2738","2739","2740","2741","2742","2743","2744","2745","2746","2747","2748","2749","2750","2751","2752","2753","2754","2755","2756","2757","2758","2759","2760","2761","2762","2763","2764","2765","2766","2767","2768","2769","2770","2771","2772","2773","2774","2775","2776","2777","2778","2779","2780","2781","2782","2783","2784","2785","2786","2787","2788","2789","2790","2791","2792","2793","2794","2795","2796","2797","2798","2799","2800","2801","2802","2803","2804","2805","2806","2807","2808","2809","2810","2811","2812","2813","2814","2815","2816","2817","2818","2819","2820","2821","2822","2823","2824","2825","2826","2827","2828","2829","2830","2831","2832","2833","2834","2835","2836","2837","2838","2839","2840","2841","2842","2843","2844","2845","2846","2847","2848","2849","2850","2851","2852","2853","2854","2855","2856","2857","2858","2859","2860","2861","2862","2863","2864","2865","2866","2867","2868","2869","2870","2871","2872","2873","2874","2875","2876","2877","2878","2879","2880","2881","2882","2883","2884","2885","2886","2887","2888","2889","2890","2891","2892","2893","2894","2895","2896","2897","2898","2899","2900","2901","2902","2903","2904","2905","2906","2907","2908","2909","2910","2911","2912","2913","2914","2915","2916","2917","2918","2919","2920","2921","2922","2923","2924","2925","2926","2927","2928","2929","2930","2931","2932","2933","2934","2935","2936","2937","2938","2939","2940","2941","2942","2943","2944","2945","2946","2947","2948","2949","2950","2951","2952","2953","2954","2955","2956","2957","2958","2959","2960","2961","2962","2963","2964","2965","2966","2967","2968","2969","2970","2971","2972","2973","2974","2975","2976","2977","2978","2979","2980","2981","2982","2983","2984","2985","2986","2987","2988","2989","2990","2991","2992","2993","2994","2995","2996","2997","2998","2999","3000","3001","3002","3003","3004","3005","3006","3007","3008","3009","3010","3011","3012","3013","3014","3015","3016","3017","3018","3019","3020","3021","3022","3023","3024","3025","3026","3027","3028","3029","3030","3031","3032","3033","3034","3035","3036","3037","3038","3039","3040","3041","3042","3043","3044","3045","3046","3047","3048","3049","3050","3051","3052","3053","3054","3055","3056","3057","3058","3059","3060","3061","3062","3063","3064","3065","3066","3067","3068","3069","3070","3071","3072","3073","3074","3075","3076","3077","3078","3079","3080","3081","3082","3083","3084","3085","3086","3087","3088","3089","3090","3091","3092","3093","3094","3095","3096","3097","3098","3099","3100","3101","3102","3103","3104","3105","3106","3107","3108","3109","3110","3111","3112","3113","3114","3115","3116","3117","3118","3119","3120","3121","3122","3123","3124","3125","3126","3127","3128","3129","3130","3131","3132","3133","3134","3135","3136","3137","3138","3139","3140","3141","3142","3143","3144","3145","3146","3147","3148","3149","3150","3151","3152","3153","3154","3155","3156","3157","3158","3159","3160","3161","3162","3163","3164","3165","3166","3167","3168","3169","3170","3171","3172","3173","3174","3175","3176","3177","3178","3179","3180","3181","3182","3183","3184","3185","3186","3187","3188","3189","3190","3191","3192","3193","3194","3195","3196","3197","3198","3199","3200","3201","3202","3203","3204","3205","3206","3207","3208","3209","3210","3211","3212","3213","3214","3215","3216","3217","3218","3219","3220","3221","3222","3223","3224","3225","3226","3227","3228","3229","3230","3231","3232","3233","3234","3235","3236","3237","3238","3239","3240","3241","3242","3243","3244","3245","3246","3247","3248","3249","3250","3251","3252","3253","3254","3255","3256","3257","3258","3259","3260","3261","3262","3263","3264","3265","3266","3267","3268","3269","3270","3271","3272","3273","3274","3275","3276","3277","3278","3279","3280","3281","3282","3283","3284","3285","3286","3287","3288","3289","3290","3291","3292","3293","3294","3295","3296","3297","3298","3299","3300","3301","3302","3303","3304","3305","3306","3307","3308","3309","3310","3311","3312","3313","3314","3315","3316","3317","3318","3319","3320","3321","3322","3323","3324","3325","3326","3327","3328","3329","3330","3331","3332","3333","3334","3335","3336","3337","3338","3339","3340","3341","3342","3343","3344","3345","3346","3347","3348","3349","3350","3351","3352","3353","3354","3355","3356","3357","3358","3359","3360","3361","3362","3363","3364","3365","3366","3367","3368","3369","3370","3371","3372","3373","3374","3375","3376","3377","3378","3379","3380","3381","3382","3383","3384","3385","3386","3387","3388","3389","3390","3391","3392","3393","3394","3395","3396","3397","3398","3399","3400","3401","3402","3403","3404","3405","3406","3407","3408","3409","3410","3411","3412","3413","3414","3415","3416","3417","3418","3419","3420","3421","3422","3423","3424","3425","3426","3427","3428","3429","3430","3431","3432","3433","3434","3435","3436","3437","3438","3439","3440","3441","3442","3443","3444","3445","3446","3447","3448","3449","3450","3451","3452","3453","3454","3455","3456","3457","3458","3459","3460","3461","3462","3463","3464","3465","3466","3467","3468","3469","3470","3471","3472","3473","3474","3475","3476","3477","3478","3479","3480","3481","3482","3483","3484","3485","3486","3487","3488","3489","3490","3491","3492","3493","3494","3495","3496","3497","3498","3499","3500","3501","3502","3503","3504","3505","3506","3507","3508","3509","3510","3511","3512","3513","3514","3515","3516","3517","3518","3519","3520","3521","3522","3523","3524","3525","3526","3527","3528","3529","3530","3531","3532","3533","3534","3535","3536","3537","3538","3539","3540","3541","3542","3543","3544","3545","3546","3547","3548","3549","3550","3551","3552","3553","3554","3555","3556","3557","3558","3559","3560","3561","3562","3563","3564","3565","3566","3567","3568","3569","3570","3571","3572","3573","3574","3575","3576","3577","3578","3579","3580","3581","3582","3583","3584","3585","3586","3587","3588","3589","3590","3591","3592","3593","3594","3595","3596","3597","3598","3599","3600","3601","3602","3603","3604","3605","3606","3607","3608","3609","3610","3611","3612","3613","3614","3615","3616","3617","3618","3619","3620","3621","3622","3623","3624","3625","3626","3627","3628","3629","3630","3631","3632","3633","3634","3635","3636","3637","3638","3639","3640","3641","3642","3643","3644","3645","3646","3647","3648","3649","3650","3651","3652","3653","3654","3655","3656","3657","3658","3659","3660","3661","3662","3663","3664","3665","3666","3667","3668","3669","3670","3671","3672","3673","3674","3675","3676","3677","3678","3679","3680","3681","3682","3683","3684","3685","3686","3687","3688","3689","3690","3691","3692","3693","3694","3695","3696","3697","3698","3699","3700","3701","3702","3703","3704","3705","3706","3707","3708","3709","3710","3711","3712","3713","3714","3715","3716","3717","3718","3719","3720","3721","3722","3723","3724","3725","3726","3727","3728","3729","3730","3731","3732","3733","3734","3735","3736","3737","3738","3739","3740","3741","3742","3743","3744","3745","3746","3747","3748","3749","3750","3751","3752","3753","3754","3755","3756","3757","3758","3759","3760","3761","3762","3763","3764","3765","3766","3767","3768","3769","3770","3771","3772","3773","3774","3775","3776","3777","3778","3779","3780","3781","3782","3783","3784","3785","3786","3787","3788","3789","3790","3791","3792","3793","3794","3795","3796","3797","3798","3799","3800","3801","3802","3803","3804","3805","3806","3807","3808","3809","3810","3811","3812","3813","3814","3815","3816","3817","3818","3819","3820","3821","3822","3823","3824","3825","3826","3827","3828","3829","3830","3831","3832","3833","3834","3835","3836","3837","3838","3839","3840","3841","3842","3843","3844","3845","3846","3847","3848","3849","3850","3851","3852","3853","3854","3855","3856","3857","3858","3859","3860","3861","3862","3863","3864","3865","3866","3867","3868","3869","3870","3871","3872","3873","3874","3875","3876","3877","3878","3879","3880","3881","3882","3883","3884","3885","3886","3887","3888","3889","3890","3891","3892","3893","3894","3895","3896","3897","3898","3899","3900","3901","3902","3903","3904","3905","3906","3907","3908","3909","3910","3911","3912","3913","3914","3915","3916","3917","3918","3919","3920","3921","3922","3923","3924","3925","3926","3927","3928","3929","3930","3931","3932","3933","3934","3935","3936","3937","3938","3939","3940","3941","3942","3943","3944","3945","3946","3947","3948","3949","3950","3951","3952","3953","3954","3955","3956","3957","3958","3959","3960","3961","3962","3963","3964","3965","3966","3967","3968","3969","3970","3971","3972","3973","3974","3975","3976","3977","3978","3979","3980","3981","3982","3983","3984","3985","3986","3987","3988","3989","3990","3991","3992","3993","3994","3995","3996","3997","3998","3999","4000","4001","4002","4003","4004","4005","4006","4007","4008","4009","4010","4011","4012","4013","4014","4015","4016","4017","4018","4019","4020","4021","4022","4023","4024","4025","4026","4027","4028","4029","4030","4031","4032","4033","4034","4035","4036","4037","4038","4039","4040","4041","4042","4043","4044","4045","4046","4047","4048","4049","4050","4051","4052","4053","4054","4055","4056","4057","4058","4059","4060","4061","4062","4063","4064","4065","4066","4067","4068","4069","4070","4071","4072","4073","4074","4075","4076","4077","4078","4079","4080","4081","4082","4083","4084","4085","4086","4087","4088","4089","4090","4091","4092","4093","4094","4095","4096","4097","4098","4099","4100","4101","4102","4103","4104","4105","4106","4107","4108","4109","4110","4111","4112","4113","4114","4115","4116","4117","4118","4119","4120","4121","4122","4123","4124","4125","4126","4127","4128","4129","4130","4131","4132","4133","4134","4135","4136","4137","4138","4139","4140","4141","4142","4143","4144","4145","4146","4147","4148","4149","4150","4151","4152","4153","4154","4155","4156","4157","4158","4159","4160","4161","4162","4163","4164","4165","4166","4167","4168","4169","4170","4171","4172","4173","4174","4175","4176","4177","4178","4179","4180","4181","4182","4183","4184","4185","4186","4187","4188","4189","4190","4191","4192","4193","4194","4195","4196","4197","4198","4199","4200","4201","4202","4203","4204","4205","4206","4207","4208","4209","4210","4211","4212","4213","4214","4215","4216","4217","4218","4219","4220","4221","4222","4223","4224","4225","4226","4227","4228","4229","4230","4231","4232","4233","4234","4235","4236","4237","4238","4239","4240","4241","4242","4243","4244","4245","4246","4247","4248","4249","4250","4251","4252","4253","4254","4255","4256","4257","4258","4259","4260","4261","4262","4263","4264","4265","4266","4267","4268","4269","4270","4271","4272","4273","4274","4275","4276","4277","4278","4279","4280","4281","4282","4283","4284","4285","4286","4287","4288","4289","4290","4291","4292","4293","4294","4295","4296","4297","4298","4299","4300","4301","4302","4303","4304","4305","4306","4307","4308","4309","4310","4311","4312","4313","4314","4315","4316","4317","4318","4319","4320","4321","4322","4323","4324","4325","4326","4327","4328","4329","4330","4331","4332","4333","4334","4335","4336","4337","4338","4339","4340","4341","4342","4343","4344","4345","4346","4347","4348","4349","4350","4351","4352","4353","4354","4355","4356","4357","4358","4359","4360","4361","4362","4363","4364","4365","4366","4367","4368","4369","4370","4371","4372","4373","4374","4375","4376","4377","4378","4379","4380","4381","4382","4383","4384","4385","4386","4387","4388","4389","4390","4391","4392","4393","4394","4395","4396","4397","4398","4399","4400","4401","4402","4403","4404","4405","4406","4407","4408","4409","4410","4411","4412","4413","4414","4415","4416","4417","4418","4419","4420","4421","4422","4423","4424","4425","4426","4427","4428","4429","4430","4431","4432","4433","4434","4435","4436","4437","4438","4439","4440","4441","4442","4443","4444","4445","4446","4447","4448","4449","4450","4451","4452","4453","4454","4455","4456","4457","4458","4459","4460","4461","4462","4463","4464","4465","4466","4467","4468","4469","4470","4471","4472","4473","4474","4475","4476","4477","4478","4479","4480","4481","4482","4483","4484","4485","4486","4487","4488","4489","4490","4491","4492","4493","4494","4495","4496","4497","4498","4499","4500","4501","4502","4503","4504","4505","4506","4507","4508","4509","4510","4511","4512","4513","4514","4515","4516","4517","4518","4519","4520","4521","4522","4523","4524","4525","4526","4527","4528","4529","4530","4531","4532","4533","4534","4535","4536","4537","4538","4539","4540","4541","4542","4543","4544","4545","4546","4547","4548","4549","4550","4551","4552","4553","4554","4555","4556","4557","4558","4559","4560","4561","4562","4563","4564","4565","4566","4567","4568","4569","4570","4571","4572","4573","4574","4575","4576","4577","4578","4579","4580","4581","4582","4583","4584","4585","4586","4587","4588","4589","4590","4591","4592","4593","4594","4595","4596","4597","4598","4599","4600","4601","4602","4603","4604","4605","4606","4607","4608","4609","4610","4611","4612","4613","4614","4615","4616","4617","4618","4619","4620","4621","4622","4623","4624","4625","4626","4627","4628","4629","4630","4631","4632","4633","4634","4635","4636","4637","4638","4639","4640","4641","4642","4643","4644","4645","4646","4647","4648","4649","4650","4651","4652","4653","4654","4655","4656","4657","4658","4659","4660","4661","4662","4663","4664","4665","4666","4667","4668","4669","4670","4671","4672","4673","4674","4675","4676","4677","4678","4679","4680","4681","4682","4683","4684","4685","4686","4687","4688","4689","4690","4691","4692","4693","4694","4695","4696","4697","4698","4699","4700","4701","4702","4703","4704","4705","4706","4707","4708","4709","4710","4711","4712","4713","4714","4715","4716","4717","4718","4719","4720","4721","4722","4723","4724","4725","4726","4727","4728","4729","4730","4731","4732","4733","4734","4735","4736","4737","4738","4739","4740","4741","4742","4743","4744","4745","4746","4747","4748","4749","4750","4751","4752","4753","4754","4755","4756","4757","4758","4759","4760","4761","4762","4763","4764","4765","4766","4767","4768","4769","4770","4771","4772","4773","4774","4775","4776","4777","4778","4779","4780","4781","4782","4783","4784","4785","4786","4787","4788","4789","4790","4791","4792","4793","4794","4795","4796","4797","4798","4799","4800","4801","4802","4803","4804","4805","4806","4807","4808","4809","4810","4811","4812","4813","4814","4815","4816","4817","4818","4819","4820","4821","4822","4823","4824","4825","4826","4827","4828","4829","4830","4831","4832","4833","4834","4835","4836","4837","4838","4839","4840","4841","4842","4843","4844","4845","4846","4847","4848","4849","4850","4851","4852","4853","4854","4855","4856","4857","4858","4859","4860","4861","4862","4863","4864","4865","4866","4867","4868","4869","4870","4871","4872","4873","4874","4875","4876","4877","4878","4879","4880","4881","4882","4883","4884","4885","4886","4887","4888","4889","4890","4891","4892","4893","4894","4895","4896","4897","4898","4899","4900","4901","4902","4903","4904","4905","4906","4907","4908","4909","4910","4911","4912","4913","4914","4915","4916","4917","4918","4919","4920","4921","4922","4923","4924","4925","4926","4927","4928","4929","4930","4931","4932","4933","4934","4935","4936","4937","4938","4939","4940","4941","4942","4943","4944","4945","4946","4947","4948","4949","4950","4951","4952","4953","4954","4955","4956","4957","4958","4959","4960","4961","4962","4963","4964","4965","4966","4967","4968","4969","4970","4971","4972","4973","4974","4975","4976","4977","4978","4979","4980","4981","4982","4983","4984","4985","4986","4987","4988","4989","4990","4991","4992","4993","4994","4995","4996","4997","4998","4999","5000","5001","5002","5003","5004","5005","5006","5007","5008","5009","5010","5011","5012","5013","5014","5015","5016","5017","5018","5019","5020","5021","5022","5023","5024","5025","5026","5027","5028","5029","5030","5031","5032","5033","5034","5035","5036","5037","5038","5039","5040","5041","5042","5043","5044","5045","5046","5047","5048","5049","5050","5051","5052","5053","5054","5055","5056","5057","5058","5059","5060","5061","5062","5063","5064","5065","5066","5067","5068","5069","5070","5071","5072","5073","5074","5075","5076","5077","5078","5079","5080","5081","5082","5083","5084","5085","5086","5087","5088","5089","5090","5091","5092","5093","5094","5095","5096","5097","5098","5099","5100","5101","5102","5103","5104","5105","5106","5107","5108","5109","5110","5111","5112","5113","5114","5115","5116","5117","5118","5119","5120","5121","5122","5123","5124","5125","5126","5127","5128","5129","5130","5131","5132","5133","5134","5135","5136","5137","5138","5139","5140","5141","5142","5143","5144","5145","5146","5147","5148","5149","5150","5151","5152","5153","5154","5155","5156","5157","5158","5159","5160","5161","5162","5163","5164","5165","5166","5167","5168","5169","5170","5171","5172","5173","5174","5175","5176","5177","5178","5179","5180","5181","5182","5183","5184","5185","5186","5187","5188","5189","5190","5191","5192","5193","5194","5195","5196","5197","5198","5199","5200","5201","5202","5203","5204","5205","5206","5207","5208","5209","5210","5211","5212","5213","5214","5215","5216","5217","5218","5219","5220","5221","5222","5223","5224","5225","5226","5227","5228","5229","5230","5231","5232","5233","5234","5235","5236","5237","5238","5239","5240","5241","5242","5243","5244","5245","5246","5247","5248","5249","5250","5251","5252","5253","5254","5255","5256","5257","5258","5259","5260","5261","5262","5263","5264","5265","5266","5267","5268","5269","5270","5271","5272","5273","5274","5275","5276","5277","5278","5279","5280","5281","5282","5283","5284","5285","5286","5287","5288","5289","5290","5291","5292","5293","5294","5295","5296","5297","5298","5299","5300","5301","5302","5303","5304","5305","5306","5307","5308","5309","5310","5311","5312","5313","5314","5315","5316","5317","5318","5319","5320","5321","5322","5323","5324","5325","5326","5327","5328","5329","5330","5331","5332","5333","5334","5335","5336","5337","5338","5339","5340","5341","5342","5343","5344","5345","5346","5347","5348","5349","5350","5351","5352","5353","5354","5355","5356","5357","5358","5359","5360","5361","5362","5363","5364","5365","5366","5367","5368","5369","5370","5371","5372","5373","5374","5375","5376","5377","5378","5379","5380","5381","5382","5383","5384","5385","5386","5387","5388","5389","5390","5391","5392","5393","5394","5395","5396","5397","5398","5399","5400","5401","5402","5403","5404","5405","5406","5407","5408","5409","5410","5411","5412","5413","5414","5415","5416","5417","5418","5419","5420","5421","5422","5423","5424","5425","5426","5427","5428","5429","5430","5431","5432","5433","5434","5435","5436","5437","5438","5439","5440","5441","5442","5443","5444","5445","5446","5447","5448","5449","5450","5451","5452","5453","5454","5455","5456","5457","5458","5459","5460","5461","5462","5463","5464","5465","5466","5467","5468","5469","5470","5471","5472","5473","5474","5475","5476","5477","5478","5479","5480","5481","5482","5483","5484","5485","5486","5487","5488","5489","5490","5491","5492","5493","5494","5495","5496","5497","5498","5499","5500","5501","5502","5503","5504","5505","5506","5507","5508","5509","5510","5511","5512","5513","5514","5515","5516","5517","5518","5519","5520","5521","5522","5523","5524","5525","5526","5527","5528","5529","5530","5531","5532","5533","5534","5535","5536","5537","5538","5539","5540","5541","5542","5543","5544","5545","5546","5547","5548","5549","5550","5551","5552","5553","5554","5555","5556","5557","5558","5559","5560","5561","5562","5563","5564","5565","5566","5567","5568","5569","5570","5571","5572","5573","5574","5575","5576","5577","5578","5579","5580","5581","5582","5583","5584","5585","5586","5587","5588","5589","5590","5591","5592","5593","5594","5595","5596","5597","5598","5599","5600","5601","5602","5603","5604","5605","5606","5607","5608","5609","5610","5611","5612","5613","5614","5615","5616","5617","5618","5619","5620","5621","5622","5623","5624","5625","5626","5627","5628","5629","5630","5631","5632","5633","5634","5635","5636","5637","5638","5639","5640","5641","5642","5643","5644","5645","5646","5647","5648","5649","5650","5651","5652","5653","5654","5655","5656","5657","5658","5659","5660","5661","5662","5663","5664","5665","5666","5667","5668","5669","5670","5671","5672","5673","5674","5675","5676","5677","5678","5679","5680","5681","5682","5683","5684","5685","5686","5687","5688","5689","5690","5691","5692","5693","5694","5695","5696","5697","5698","5699","5700","5701","5702","5703","5704","5705","5706","5707","5708","5709","5710","5711","5712","5713","5714","5715","5716","5717","5718","5719","5720","5721","5722","5723","5724","5725","5726","5727","5728","5729","5730","5731","5732","5733","5734","5735","5736","5737","5738","5739","5740","5741","5742","5743","5744","5745","5746","5747","5748","5749","5750","5751","5752","5753","5754","5755","5756","5757","5758","5759","5760","5761","5762","5763","5764","5765","5766","5767","5768","5769","5770","5771","5772","5773","5774","5775","5776","5777","5778","5779","5780","5781","5782","5783","5784","5785","5786","5787","5788","5789","5790","5791","5792","5793","5794","5795","5796","5797","5798","5799","5800","5801","5802","5803","5804","5805","5806","5807","5808","5809","5810","5811","5812","5813","5814","5815","5816","5817","5818","5819","5820","5821","5822","5823","5824","5825","5826","5827","5828","5829","5830","5831","5832","5833","5834","5835","5836","5837","5838","5839","5840","5841","5842","5843","5844","5845","5846","5847","5848","5849","5850","5851","5852","5853","5854","5855","5856","5857","5858","5859","5860","5861","5862","5863","5864","5865","5866","5867","5868","5869","5870","5871","5872","5873","5874","5875","5876","5877","5878","5879","5880","5881","5882","5883","5884","5885","5886","5887","5888","5889","5890","5891","5892","5893","5894","5895","5896","5897","5898","5899","5900","5901","5902","5903","5904","5905","5906","5907","5908","5909","5910","5911","5912","5913","5914","5915","5916","5917","5918","5919","5920","5921","5922","5923","5924","5925","5926","5927","5928","5929","5930","5931","5932","5933","5934","5935","5936","5937","5938","5939","5940","5941","5942","5943","5944","5945","5946","5947","5948","5949","5950","5951","5952","5953","5954","5955","5956","5957","5958","5959","5960","5961","5962","5963","5964","5965","5966","5967","5968","5969","5970","5971","5972","5973","5974","5975","5976","5977","5978","5979","5980","5981","5982","5983","5984","5985","5986","5987","5988","5989","5990","5991","5992","5993","5994","5995","5996","5997","5998","5999","6000","6001","6002","6003","6004","6005","6006","6007","6008","6009","6010","6011","6012","6013","6014","6015","6016","6017","6018","6019","6020","6021","6022","6023","6024","6025","6026","6027","6028","6029","6030","6031","6032","6033","6034","6035","6036","6037","6038","6039","6040","6041","6042","6043","6044","6045","6046","6047","6048","6049","6050","6051","6052","6053","6054","6055","6056","6057","6058","6059","6060","6061","6062","6063","6064","6065","6066","6067","6068","6069","6070","6071","6072","6073","6074","6075","6076","6077","6078","6079","6080","6081","6082","6083","6084","6085","6086","6087","6088","6089","6090","6091","6092","6093","6094","6095","6096","6097","6098","6099","6100","6101","6102","6103","6104","6105","6106","6107","6108","6109","6110","6111","6112","6113","6114","6115","6116","6117","6118","6119","6120","6121","6122","6123","6124","6125","6126","6127","6128","6129","6130","6131","6132","6133","6134","6135","6136","6137","6138","6139","6140","6141","6142","6143","6144","6145","6146","6147","6148","6149","6150","6151","6152","6153","6154","6155","6156","6157","6158","6159","6160","6161","6162","6163","6164","6165","6166","6167","6168","6169","6170","6171","6172","6173","6174","6175","6176","6177","6178","6179","6180","6181","6182","6183","6184","6185","6186","6187","6188","6189","6190","6191","6192","6193","6194","6195","6196","6197","6198","6199","6200","6201","6202","6203","6204","6205","6206","6207","6208","6209","6210","6211","6212","6213","6214","6215","6216","6217","6218","6219","6220","6221","6222","6223","6224","6225","6226","6227","6228","6229","6230","6231","6232","6233","6234","6235","6236","6237","6238","6239","6240","6241","6242","6243","6244","6245","6246","6247","6248","6249","6250","6251","6252","6253","6254","6255","6256","6257","6258","6259","6260","6261","6262","6263","6264","6265","6266","6267","6268","6269","6270","6271","6272","6273","6274","6275","6276","6277","6278","6279","6280","6281","6282","6283","6284","6285","6286","6287","6288","6289","6290","6291","6292","6293","6294","6295","6296","6297","6298","6299","6300","6301","6302","6303","6304","6305","6306","6307","6308","6309","6310","6311","6312","6313","6314","6315","6316","6317","6318","6319","6320","6321","6322","6323","6324","6325","6326","6327","6328","6329","6330","6331","6332","6333","6334","6335","6336","6337","6338","6339","6340","6341","6342","6343","6344","6345","6346","6347","6348","6349","6350","6351","6352","6353","6354","6355","6356","6357","6358","6359","6360","6361","6362","6363","6364","6365","6366","6367","6368","6369","6370","6371","6372","6373","6374","6375","6376","6377","6378","6379","6380","6381","6382","6383","6384","6385","6386","6387","6388","6389","6390","6391","6392","6393","6394","6395","6396","6397","6398","6399","6400","6401","6402","6403","6404","6405","6406","6407","6408","6409","6410","6411","6412","6413","6414","6415","6416","6417","6418","6419","6420","6421","6422","6423","6424","6425","6426","6427","6428","6429","6430","6431","6432","6433","6434","6435","6436","6437","6438","6439","6440","6441","6442","6443","6444","6445","6446","6447","6448","6449","6450","6451","6452","6453","6454","6455","6456","6457","6458","6459","6460","6461","6462","6463","6464","6465","6466","6467","6468","6469","6470","6471","6472","6473","6474","6475","6476","6477","6478","6479","6480","6481","6482","6483","6484","6485","6486","6487","6488","6489","6490","6491","6492","6493","6494","6495","6496","6497","6498","6499","6500","6501","6502","6503","6504","6505","6506","6507","6508","6509","6510","6511","6512","6513","6514","6515","6516","6517","6518","6519","6520","6521","6522","6523","6524","6525","6526","6527","6528","6529","6530","6531","6532","6533","6534","6535","6536","6537","6538","6539","6540","6541","6542","6543","6544","6545","6546","6547","6548","6549","6550","6551","6552","6553","6554","6555","6556","6557","6558","6559","6560","6561","6562","6563","6564","6565","6566","6567","6568","6569","6570","6571","6572","6573","6574","6575","6576","6577","6578","6579","6580","6581","6582","6583","6584","6585","6586","6587","6588","6589","6590","6591","6592","6593","6594","6595","6596","6597","6598","6599","6600","6601","6602","6603","6604","6605","6606","6607","6608","6609","6610","6611","6612","6613","6614","6615","6616","6617","6618","6619","6620","6621","6622","6623","6624","6625","6626","6627","6628","6629","6630","6631","6632","6633","6634","6635","6636","6637","6638","6639","6640","6641","6642","6643","6644","6645","6646","6647","6648","6649","6650","6651","6652","6653","6654","6655","6656","6657","6658","6659","6660","6661","6662","6663","6664","6665","6666","6667","6668","6669","6670","6671","6672","6673","6674","6675","6676","6677","6678","6679","6680","6681","6682","6683","6684","6685","6686","6687","6688","6689","6690","6691","6692","6693","6694","6695","6696","6697","6698","6699","6700","6701","6702","6703","6704","6705","6706","6707","6708","6709","6710","6711","6712","6713","6714","6715","6716","6717","6718","6719","6720","6721","6722","6723","6724","6725","6726","6727","6728","6729","6730","6731","6732","6733","6734","6735","6736","6737","6738","6739","6740","6741","6742","6743","6744","6745","6746","6747","6748","6749","6750","6751","6752","6753","6754","6755","6756","6757","6758","6759","6760","6761","6762","6763","6764","6765","6766","6767","6768","6769","6770","6771","6772","6773","6774","6775","6776","6777","6778","6779","6780","6781","6782","6783","6784","6785","6786","6787","6788","6789","6790","6791","6792","6793","6794","6795","6796","6797","6798","6799","6800","6801","6802","6803","6804","6805","6806","6807","6808","6809","6810","6811","6812","6813","6814","6815","6816","6817","6818","6819","6820","6821","6822","6823","6824","6825","6826","6827","6828","6829","6830","6831","6832","6833","6834","6835","6836","6837","6838","6839","6840","6841","6842","6843","6844","6845","6846","6847","6848","6849","6850","6851","6852","6853","6854","6855","6856","6857","6858","6859","6860","6861","6862","6863","6864","6865","6866","6867","6868","6869","6870","6871","6872","6873","6874","6875","6876","6877","6878","6879","6880","6881","6882","6883","6884","6885","6886","6887","6888","6889","6890","6891","6892","6893","6894","6895","6896","6897","6898","6899","6900","6901","6902","6903","6904","6905","6906","6907","6908","6909","6910","6911","6912","6913","6914","6915","6916","6917","6918","6919","6920","6921","6922","6923","6924","6925","6926","6927","6928","6929","6930","6931","6932","6933","6934","6935","6936","6937","6938","6939","6940","6941","6942","6943","6944","6945","6946","6947","6948","6949","6950","6951","6952","6953","6954","6955","6956","6957","6958","6959","6960","6961","6962","6963","6964","6965","6966","6967","6968","6969","6970","6971","6972","6973","6974","6975","6976","6977","6978","6979","6980","6981","6982","6983","6984","6985","6986","6987","6988","6989","6990","6991","6992","6993","6994","6995","6996","6997","6998","6999","7000","7001","7002","7003","7004","7005","7006","7007","7008","7009","7010","7011","7012","7013","7014","7015","7016","7017","7018","7019","7020","7021","7022","7023","7024","7025","7026","7027","7028","7029","7030","7031","7032","7033","7034","7035","7036","7037","7038","7039","7040","7041","7042","7043","7044","7045","7046","7047","7048","7049","7050","7051","7052","7053","7054","7055","7056","7057","7058","7059","7060","7061","7062","7063","7064","7065","7066","7067","7068","7069","7070","7071","7072","7073","7074","7075","7076","7077","7078","7079","7080","7081","7082","7083","7084","7085","7086","7087","7088","7089","7090","7091","7092","7093","7094","7095","7096","7097","7098","7099","7100","7101","7102","7103","7104","7105","7106","7107","7108","7109","7110","7111","7112","7113","7114","7115","7116","7117","7118","7119","7120","7121","7122","7123","7124","7125","7126","7127","7128","7129","7130","7131","7132","7133","7134","7135","7136","7137","7138","7139","7140","7141","7142","7143","7144","7145","7146","7147","7148","7149","7150","7151","7152","7153","7154","7155","7156","7157","7158","7159","7160","7161","7162","7163","7164","7165","7166","7167","7168","7169","7170","7171","7172","7173","7174","7175","7176","7177","7178","7179","7180","7181","7182","7183","7184","7185","7186","7187","7188","7189","7190","7191","7192","7193","7194","7195","7196","7197","7198","7199","7200","7201","7202","7203","7204","7205","7206","7207","7208","7209","7210","7211","7212","7213","7214","7215","7216","7217","7218","7219","7220","7221","7222","7223","7224","7225","7226","7227","7228","7229","7230","7231","7232","7233","7234","7235","7236","7237","7238","7239","7240","7241","7242","7243","7244","7245","7246","7247","7248","7249","7250","7251","7252","7253","7254","7255","7256","7257","7258","7259","7260","7261","7262","7263","7264","7265","7266","7267","7268","7269","7270","7271","7272","7273","7274","7275","7276","7277","7278","7279","7280","7281","7282","7283","7284","7285","7286","7287","7288","7289","7290","7291","7292","7293","7294","7295","7296","7297","7298","7299","7300","7301","7302","7303","7304","7305","7306","7307","7308","7309","7310","7311","7312","7313","7314","7315","7316","7317","7318","7319","7320","7321","7322","7323","7324","7325","7326","7327","7328","7329","7330","7331","7332","7333","7334","7335","7336","7337","7338","7339","7340","7341","7342","7343","7344","7345","7346","7347","7348","7349","7350","7351","7352","7353","7354","7355","7356","7357","7358","7359","7360","7361","7362","7363","7364","7365","7366","7367","7368","7369","7370","7371","7372","7373","7374","7375","7376","7377","7378","7379","7380","7381","7382","7383","7384","7385","7386","7387","7388","7389","7390","7391","7392","7393","7394","7395","7396","7397","7398","7399","7400","7401","7402","7403","7404","7405","7406","7407","7408","7409","7410","7411","7412","7413","7414","7415","7416","7417","7418","7419","7420","7421","7422","7423","7424","7425","7426","7427","7428","7429","7430","7431","7432","7433","7434","7435","7436","7437","7438","7439","7440","7441","7442","7443","7444","7445","7446","7447","7448","7449","7450","7451","7452","7453","7454","7455","7456","7457","7458","7459","7460","7461","7462","7463","7464","7465","7466","7467","7468","7469","7470","7471","7472","7473","7474","7475","7476","7477","7478","7479","7480","7481","7482","7483","7484","7485","7486","7487","7488","7489","7490","7491","7492","7493","7494","7495","7496","7497","7498","7499","7500","7501","7502","7503","7504","7505","7506","7507","7508","7509","7510","7511","7512","7513","7514","7515","7516","7517","7518","7519","7520","7521","7522","7523","7524","7525","7526","7527","7528","7529","7530","7531","7532","7533","7534","7535","7536","7537","7538","7539","7540","7541","7542","7543","7544","7545","7546","7547","7548","7549","7550","7551","7552","7553","7554","7555","7556","7557","7558","7559","7560","7561","7562","7563","7564","7565","7566","7567","7568","7569","7570","7571","7572","7573","7574","7575","7576","7577","7578","7579","7580","7581","7582","7583","7584","7585","7586","7587","7588","7589","7590","7591","7592","7593","7594","7595","7596","7597","7598","7599","7600","7601","7602","7603","7604","7605","7606","7607","7608","7609","7610","7611","7612","7613","7614","7615","7616","7617","7618","7619","7620","7621","7622","7623","7624","7625","7626","7627","7628","7629","7630","7631","7632","7633","7634","7635","7636","7637","7638","7639","7640","7641","7642","7643","7644","7645","7646","7647","7648","7649","7650","7651","7652","7653","7654","7655","7656","7657","7658","7659","7660","7661","7662","7663","7664","7665","7666","7667","7668","7669","7670","7671","7672","7673","7674","7675","7676","7677","7678","7679","7680","7681","7682","7683","7684","7685","7686","7687","7688","7689","7690","7691","7692","7693","7694","7695","7696","7697","7698","7699","7700","7701","7702","7703","7704","7705","7706","7707","7708","7709","7710","7711","7712","7713","7714","7715","7716","7717","7718","7719","7720","7721","7722","7723","7724","7725","7726","7727","7728","7729","7730","7731","7732","7733","7734","7735","7736","7737","7738","7739","7740","7741","7742","7743","7744","7745","7746","7747","7748","7749","7750","7751","7752","7753","7754","7755","7756","7757","7758","7759","7760","7761","7762","7763","7764","7765","7766","7767","7768","7769","7770","7771","7772","7773","7774","7775","7776","7777","7778","7779","7780","7781","7782","7783","7784","7785","7786","7787","7788","7789","7790","7791","7792","7793","7794","7795","7796","7797","7798","7799","7800","7801","7802","7803","7804","7805","7806","7807","7808","7809","7810","7811","7812","7813","7814","7815","7816","7817","7818","7819","7820","7821","7822","7823","7824","7825","7826","7827","7828","7829","7830","7831","7832","7833","7834","7835","7836","7837","7838","7839","7840","7841","7842","7843","7844","7845","7846","7847","7848","7849","7850","7851","7852","7853","7854","7855","7856","7857","7858","7859","7860","7861","7862","7863","7864","7865","7866","7867","7868","7869","7870","7871","7872","7873","7874","7875","7876","7877","7878","7879","7880","7881","7882","7883","7884","7885","7886","7887","7888","7889","7890","7891","7892","7893","7894","7895","7896","7897","7898","7899","7900","7901","7902","7903","7904","7905","7906","7907","7908","7909","7910","7911","7912","7913","7914","7915","7916","7917","7918","7919","7920","7921","7922","7923","7924","7925","7926","7927","7928","7929","7930","7931","7932","7933","7934","7935","7936","7937","7938","7939","7940","7941","7942","7943","7944","7945","7946","7947","7948","7949","7950","7951","7952","7953","7954","7955","7956","7957","7958","7959","7960","7961","7962","7963","7964","7965","7966","7967","7968","7969","7970","7971","7972","7973","7974","7975","7976","7977","7978","7979","7980","7981","7982","7983","7984","7985","7986","7987","7988","7989","7990","7991","7992","7993","7994","7995","7996","7997","7998","7999","8000","8001","8002","8003","8004","8005","8006","8007","8008","8009","8010","8011","8012","8013","8014","8015","8016","8017","8018","8019","8020","8021","8022","8023","8024","8025","8026","8027","8028","8029","8030","8031","8032","8033","8034","8035","8036","8037","8038","8039","8040","8041","8042","8043","8044","8045","8046","8047","8048","8049","8050","8051","8052","8053","8054","8055","8056","8057","8058","8059","8060","8061","8062","8063","8064","8065","8066","8067","8068","8069","8070","8071","8072","8073","8074","8075","8076","8077","8078","8079","8080","8081","8082","8083","8084","8085","8086","8087","8088","8089","8090","8091","8092","8093","8094","8095","8096","8097","8098","8099","8100","8101","8102","8103","8104","8105","8106","8107","8108","8109","8110","8111","8112","8113","8114","8115","8116","8117","8118","8119","8120","8121","8122","8123","8124","8125","8126","8127","8128","8129","8130","8131","8132","8133","8134","8135","8136","8137","8138","8139","8140","8141","8142","8143","8144","8145","8146","8147","8148","8149","8150","8151","8152","8153","8154","8155","8156","8157","8158","8159","8160","8161","8162","8163","8164","8165","8166","8167","8168","8169","8170","8171","8172","8173","8174","8175","8176","8177","8178","8179","8180","8181","8182","8183","8184","8185","8186","8187","8188","8189","8190","8191","8192","8193","8194","8195","8196","8197","8198","8199","8200","8201","8202","8203","8204","8205","8206","8207","8208","8209","8210","8211","8212","8213","8214","8215","8216","8217","8218","8219","8220","8221","8222","8223","8224","8225","8226","8227","8228","8229","8230","8231","8232","8233","8234","8235","8236","8237","8238","8239","8240","8241","8242","8243","8244","8245","8246","8247","8248","8249","8250","8251","8252","8253","8254","8255","8256","8257","8258","8259","8260","8261","8262","8263","8264","8265","8266","8267","8268","8269","8270","8271","8272","8273","8274","8275","8276","8277","8278","8279","8280","8281","8282","8283","8284","8285","8286","8287","8288","8289","8290","8291","8292","8293","8294","8295","8296","8297","8298","8299","8300","8301","8302","8303","8304","8305","8306","8307","8308","8309","8310","8311","8312","8313","8314","8315","8316","8317","8318","8319","8320","8321","8322","8323","8324","8325","8326","8327","8328","8329","8330","8331","8332","8333","8334","8335","8336","8337","8338","8339","8340","8341","8342","8343","8344","8345","8346","8347","8348","8349","8350","8351","8352","8353","8354","8355","8356","8357","8358","8359","8360","8361","8362","8363","8364","8365","8366","8367","8368","8369","8370","8371","8372","8373","8374","8375","8376","8377","8378","8379","8380","8381","8382","8383","8384","8385","8386","8387","8388","8389","8390","8391","8392","8393","8394","8395","8396","8397","8398","8399","8400","8401","8402","8403","8404","8405","8406","8407","8408","8409","8410","8411","8412","8413","8414","8415","8416","8417","8418","8419","8420","8421","8422","8423","8424","8425","8426","8427","8428","8429","8430","8431","8432","8433","8434","8435","8436","8437","8438","8439","8440","8441","8442","8443","8444","8445","8446","8447","8448","8449","8450","8451","8452","8453","8454","8455","8456","8457","8458","8459","8460","8461","8462","8463","8464","8465","8466","8467","8468","8469","8470","8471","8472","8473","8474","8475","8476","8477","8478","8479","8480","8481","8482","8483","8484","8485","8486","8487","8488","8489","8490","8491","8492","8493","8494","8495","8496","8497","8498","8499","8500","8501","8502","8503","8504","8505","8506","8507","8508","8509","8510","8511","8512","8513","8514","8515","8516","8517","8518","8519","8520","8521","8522","8523","8524","8525","8526","8527","8528","8529","8530","8531","8532","8533","8534","8535","8536","8537","8538","8539","8540","8541","8542","8543","8544","8545","8546","8547","8548","8549","8550","8551","8552","8553","8554","8555","8556","8557","8558","8559","8560","8561","8562","8563","8564","8565","8566","8567","8568","8569","8570","8571","8572","8573","8574","8575","8576","8577","8578","8579","8580","8581","8582","8583","8584","8585","8586","8587","8588","8589","8590","8591","8592","8593","8594","8595","8596","8597","8598","8599","8600","8601","8602","8603","8604","8605","8606","8607","8608","8609","8610","8611","8612","8613","8614","8615","8616","8617","8618","8619","8620","8621","8622","8623","8624","8625","8626","8627","8628","8629","8630","8631","8632","8633","8634","8635","8636","8637","8638","8639","8640","8641","8642","8643","8644","8645","8646","8647","8648","8649","8650","8651","8652","8653","8654","8655","8656","8657","8658","8659","8660","8661","8662","8663","8664","8665","8666","8667","8668","8669","8670","8671","8672","8673","8674","8675","8676","8677","8678","8679","8680","8681","8682","8683","8684","8685","8686","8687","8688","8689","8690","8691","8692","8693","8694","8695","8696","8697","8698","8699","8700","8701","8702","8703","8704","8705","8706","8707","8708","8709","8710","8711","8712","8713","8714","8715","8716","8717","8718","8719","8720","8721","8722","8723","8724","8725","8726","8727","8728","8729","8730","8731","8732","8733","8734","8735","8736","8737","8738","8739","8740","8741","8742","8743","8744","8745","8746","8747","8748","8749","8750","8751","8752","8753","8754","8755","8756","8757","8758","8759","8760","8761","8762","8763","8764","8765","8766","8767","8768","8769","8770","8771","8772","8773","8774","8775","8776","8777","8778","8779","8780","8781","8782","8783","8784","8785","8786","8787","8788","8789","8790","8791","8792","8793","8794","8795","8796","8797","8798","8799","8800","8801","8802","8803","8804","8805","8806","8807","8808","8809","8810","8811","8812","8813","8814","8815","8816","8817","8818","8819","8820","8821","8822","8823","8824","8825","8826","8827","8828","8829","8830","8831","8832","8833","8834","8835","8836","8837","8838","8839","8840","8841","8842","8843","8844","8845","8846","8847","8848","8849","8850","8851","8852","8853","8854","8855","8856","8857","8858","8859","8860","8861","8862","8863","8864","8865","8866","8867","8868","8869","8870","8871","8872","8873","8874","8875","8876","8877","8878","8879","8880","8881","8882","8883","8884","8885","8886","8887","8888","8889","8890","8891","8892","8893","8894","8895","8896","8897","8898","8899","8900","8901","8902","8903","8904","8905","8906","8907","8908","8909","8910","8911","8912","8913","8914","8915","8916","8917","8918","8919","8920","8921","8922","8923","8924","8925","8926","8927","8928","8929","8930","8931","8932","8933","8934","8935","8936","8937","8938","8939","8940","8941","8942","8943","8944","8945","8946","8947","8948","8949","8950","8951","8952","8953","8954","8955","8956","8957","8958","8959","8960","8961","8962","8963","8964","8965","8966","8967","8968","8969","8970","8971","8972","8973","8974","8975","8976","8977","8978","8979","8980","8981","8982","8983","8984","8985","8986","8987","8988","8989","8990","8991","8992","8993","8994","8995","8996","8997","8998","8999","9000","9001","9002","9003","9004","9005","9006","9007","9008","9009","9010","9011","9012","9013","9014","9015","9016","9017","9018","9019","9020","9021","9022","9023","9024","9025","9026","9027","9028","9029","9030","9031","9032","9033","9034","9035","9036","9037","9038","9039","9040","9041","9042","9043","9044","9045","9046","9047","9048","9049","9050","9051","9052","9053","9054","9055","9056","9057","9058","9059","9060","9061","9062","9063","9064","9065","9066","9067","9068","9069","9070","9071","9072","9073","9074","9075","9076","9077","9078","9079","9080","9081","9082","9083","9084","9085","9086","9087","9088","9089","9090","9091","9092","9093","9094","9095","9096","9097","9098","9099","9100","9101","9102","9103","9104","9105","9106","9107","9108","9109","9110","9111","9112","9113","9114","9115","9116","9117","9118","9119","9120","9121","9122","9123","9124","9125","9126","9127","9128","9129","9130","9131","9132","9133","9134","9135","9136","9137","9138","9139","9140","9141","9142","9143","9144","9145","9146","9147","9148","9149","9150","9151","9152","9153","9154","9155","9156","9157","9158","9159","9160","9161","9162","9163","9164","9165","9166","9167","9168","9169","9170","9171","9172","9173","9174","9175","9176","9177","9178","9179","9180","9181","9182","9183","9184","9185","9186","9187","9188","9189","9190","9191","9192","9193","9194","9195","9196","9197","9198","9199","9200","9201","9202","9203","9204","9205","9206","9207","9208","9209","9210","9211","9212","9213","9214","9215","9216","9217","9218","9219","9220","9221","9222","9223","9224","9225","9226","9227","9228","9229","9230","9231","9232","9233","9234","9235","9236","9237","9238","9239","9240","9241","9242","9243","9244","9245","9246","9247","9248","9249","9250","9251","9252","9253","9254","9255","9256","9257","9258","9259","9260","9261","9262","9263","9264","9265","9266","9267","9268","9269","9270","9271","9272","9273","9274","9275","9276","9277","9278","9279","9280","9281","9282","9283","9284","9285","9286","9287","9288","9289","9290","9291","9292","9293","9294","9295","9296","9297","9298","9299","9300","9301","9302","9303","9304","9305","9306","9307","9308","9309","9310","9311","9312","9313","9314","9315","9316","9317","9318","9319","9320","9321","9322","9323","9324","9325","9326","9327","9328","9329","9330","9331","9332","9333","9334","9335","9336","9337","9338","9339","9340","9341","9342","9343","9344","9345","9346","9347","9348","9349","9350","9351","9352","9353","9354","9355","9356","9357","9358","9359","9360","9361","9362","9363","9364","9365","9366","9367","9368","9369","9370","9371","9372","9373","9374","9375","9376","9377","9378","9379","9380","9381","9382","9383","9384","9385","9386","9387","9388","9389","9390","9391","9392","9393","9394","9395","9396","9397","9398","9399","9400","9401","9402","9403","9404","9405","9406","9407","9408","9409","9410","9411","9412","9413","9414","9415","9416","9417","9418","9419","9420","9421","9422","9423","9424","9425","9426","9427","9428","9429","9430","9431","9432","9433","9434","9435","9436","9437","9438","9439","9440","9441","9442","9443","9444","9445","9446","9447","9448","9449","9450","9451","9452","9453","9454","9455","9456","9457","9458","9459","9460","9461","9462","9463","9464","9465","9466","9467","9468","9469","9470","9471","9472","9473","9474","9475","9476","9477","9478","9479","9480","9481","9482","9483","9484","9485","9486","9487","9488","9489","9490","9491","9492","9493","9494","9495","9496","9497","9498","9499","9500","9501","9502","9503","9504","9505","9506","9507","9508","9509","9510","9511","9512","9513","9514","9515","9516","9517","9518","9519","9520","9521","9522","9523","9524","9525","9526","9527","9528","9529","9530","9531","9532","9533","9534","9535","9536","9537","9538","9539","9540","9541","9542","9543","9544","9545","9546","9547","9548","9549","9550","9551","9552","9553","9554","9555","9556","9557","9558","9559","9560","9561","9562","9563","9564","9565","9566","9567","9568","9569","9570","9571","9572","9573","9574","9575","9576","9577","9578","9579","9580","9581","9582","9583","9584","9585","9586","9587","9588","9589","9590","9591","9592","9593","9594","9595","9596","9597","9598","9599","9600","9601","9602","9603","9604","9605","9606","9607","9608","9609","9610","9611","9612","9613","9614","9615","9616","9617","9618","9619","9620","9621","9622","9623","9624","9625","9626","9627","9628","9629","9630","9631","9632","9633","9634","9635","9636","9637","9638","9639","9640","9641","9642","9643","9644","9645","9646","9647","9648","9649","9650","9651","9652","9653","9654","9655","9656","9657","9658","9659","9660","9661","9662","9663","9664","9665","9666","9667","9668","9669","9670","9671","9672","9673","9674","9675","9676","9677","9678","9679","9680","9681","9682","9683","9684","9685","9686","9687","9688","9689","9690","9691","9692","9693","9694","9695","9696","9697","9698","9699","9700","9701","9702","9703","9704","9705","9706","9707","9708","9709","9710","9711","9712","9713","9714","9715","9716","9717","9718","9719","9720","9721","9722","9723","9724","9725","9726","9727","9728","9729","9730","9731","9732","9733","9734","9735","9736","9737","9738","9739","9740","9741","9742","9743","9744","9745","9746","9747","9748","9749","9750","9751","9752","9753","9754","9755","9756","9757","9758","9759","9760","9761","9762","9763","9764","9765","9766","9767","9768","9769","9770","9771","9772","9773","9774","9775","9776","9777","9778","9779","9780","9781","9782","9783","9784","9785","9786","9787","9788","9789","9790","9791","9792","9793","9794","9795","9796","9797","9798","9799","9800","9801","9802","9803","9804","9805","9806","9807","9808","9809","9810","9811","9812","9813","9814","9815","9816","9817","9818","9819","9820","9821","9822","9823","9824","9825","9826","9827","9828","9829","9830","9831","9832","9833","9834","9835","9836","9837","9838","9839","9840","9841","9842","9843","9844","9845","9846","9847","9848","9849","9850","9851","9852","9853","9854","9855","9856","9857","9858","9859","9860","9861","9862","9863","9864","9865","9866","9867","9868","9869","9870","9871","9872","9873","9874","9875","9876","9877","9878","9879","9880","9881","9882","9883","9884","9885","9886","9887","9888","9889","9890","9891","9892","9893","9894","9895","9896","9897","9898","9899","9900","9901","9902","9903","9904","9905","9906","9907","9908","9909","9910","9911","9912","9913","9914","9915","9916","9917","9918","9919","9920","9921","9922","9923","9924","9925","9926","9927","9928","9929","9930","9931","9932","9933","9934","9935","9936","9937","9938","9939","9940","9941","9942","9943","9944","9945","9946","9947","9948","9949","9950","9951","9952","9953","9954","9955","9956","9957","9958","9959","9960","9961","9962","9963","9964","9965","9966","9967","9968","9969","9970","9971","9972","9973","9974","9975","9976","9977","9978","9979","9980","9981","9982","9983","9984","9985","9986","9987","9988","9989","9990","9991","9992","9993","9994","9995","9996","9997","9998","9999","10000","10001","10002","10003","10004","10005","10006","10007","10008","10009","10010","10011","10012","10013","10014","10015","10016","10017","10018","10019","10020","10021","10022","10023","10024","10025","10026","10027","10028","10029","10030","10031","10032","10033","10034","10035","10036","10037","10038","10039","10040","10041","10042","10043","10044","10045","10046","10047","10048","10049","10050","10051","10052","10053","10054","10055","10056","10057","10058","10059","10060","10061","10062","10063","10064","10065","10066","10067","10068","10069","10070","10071","10072","10073","10074","10075","10076","10077","10078","10079","10080","10081","10082","10083","10084","10085","10086","10087","10088","10089","10090","10091","10092","10093","10094","10095","10096","10097","10098","10099","10100","10101","10102","10103","10104","10105","10106","10107","10108","10109","10110","10111","10112","10113","10114","10115","10116","10117","10118","10119","10120","10121","10122","10123","10124","10125","10126","10127","10128","10129","10130","10131","10132","10133","10134","10135","10136","10137","10138","10139","10140","10141","10142","10143","10144","10145","10146","10147","10148","10149","10150","10151","10152","10153","10154","10155","10156","10157","10158","10159","10160","10161","10162","10163","10164","10165","10166","10167","10168","10169","10170","10171","10172","10173","10174","10175","10176","10177","10178","10179","10180","10181","10182","10183","10184","10185","10186","10187","10188","10189","10190","10191","10192","10193","10194","10195","10196","10197","10198","10199","10200","10201","10202","10203","10204","10205","10206","10207","10208","10209","10210","10211","10212","10213","10214","10215","10216","10217","10218","10219","10220","10221","10222","10223","10224","10225","10226","10227","10228","10229","10230","10231","10232","10233","10234","10235","10236","10237","10238","10239","10240","10241","10242","10243","10244","10245","10246","10247","10248","10249","10250","10251","10252","10253","10254","10255","10256","10257","10258","10259","10260","10261","10262","10263","10264","10265","10266","10267","10268","10269","10270","10271","10272","10273","10274","10275","10276","10277","10278","10279","10280","10281","10282","10283","10284","10285","10286","10287","10288","10289","10290","10291","10292","10293","10294","10295","10296","10297","10298","10299","10300","10301","10302","10303","10304","10305","10306","10307","10308","10309","10310","10311","10312","10313","10314","10315","10316","10317","10318","10319","10320","10321","10322","10323","10324","10325","10326","10327","10328","10329","10330","10331","10332","10333","10334","10335","10336","10337","10338","10339","10340","10341","10342","10343","10344","10345","10346","10347","10348","10349","10350","10351","10352","10353","10354","10355","10356","10357","10358","10359","10360","10361","10362","10363","10364","10365","10366","10367","10368","10369","10370","10371","10372","10373","10374","10375","10376","10377","10378","10379","10380","10381","10382","10383","10384","10385","10386","10387","10388","10389","10390","10391","10392","10393","10394","10395","10396","10397","10398","10399","10400","10401","10402","10403","10404","10405","10406","10407","10408","10409","10410","10411","10412","10413","10414","10415","10416","10417","10418","10419","10420","10421","10422","10423","10424","10425","10426","10427","10428","10429","10430","10431","10432","10433","10434","10435","10436","10437","10438","10439","10440","10441","10442","10443","10444","10445","10446","10447","10448","10449","10450","10451","10452","10453","10454","10455","10456","10457","10458","10459","10460","10461","10462","10463","10464","10465","10466","10467","10468","10469","10470","10471","10472","10473","10474","10475","10476","10477","10478","10479","10480","10481","10482","10483","10484","10485","10486","10487","10488","10489","10490","10491","10492","10493","10494","10495","10496","10497","10498","10499","10500","10501","10502","10503","10504","10505","10506","10507","10508","10509","10510","10511","10512","10513","10514","10515","10516","10517","10518","10519","10520","10521","10522","10523","10524","10525","10526","10527","10528","10529","10530","10531","10532","10533","10534","10535","10536","10537","10538","10539","10540","10541","10542","10543","10544","10545","10546","10547","10548","10549","10550","10551","10552","10553","10554","10555","10556","10557","10558","10559","10560","10561","10562","10563","10564","10565","10566","10567","10568","10569","10570","10571","10572","10573","10574","10575","10576","10577","10578","10579","10580","10581","10582","10583","10584","10585","10586","10587","10588","10589","10590","10591","10592","10593","10594","10595","10596","10597","10598","10599","10600","10601","10602","10603","10604","10605","10606","10607","10608","10609","10610","10611","10612","10613","10614","10615","10616","10617","10618","10619","10620","10621","10622","10623","10624","10625","10626","10627","10628","10629","10630","10631","10632","10633","10634","10635","10636","10637","10638","10639","10640","10641","10642","10643","10644","10645","10646","10647","10648","10649","10650","10651","10652","10653","10654","10655","10656","10657","10658","10659","10660","10661","10662","10663","10664","10665","10666","10667","10668","10669","10670","10671","10672","10673","10674","10675","10676","10677","10678","10679","10680","10681","10682","10683","10684","10685","10686","10687","10688","10689","10690","10691","10692","10693","10694","10695","10696","10697","10698","10699","10700","10701","10702","10703","10704","10705","10706","10707","10708","10709","10710","10711","10712","10713","10714","10715","10716","10717","10718","10719","10720","10721","10722","10723","10724","10725","10726","10727","10728","10729","10730","10731","10732","10733","10734","10735","10736","10737","10738","10739","10740","10741","10742","10743","10744","10745","10746","10747","10748","10749","10750","10751","10752","10753","10754","10755","10756","10757","10758","10759","10760","10761","10762","10763","10764","10765","10766","10767","10768","10769","10770","10771","10772","10773","10774","10775","10776","10777","10778","10779","10780","10781","10782","10783","10784","10785","10786","10787","10788","10789","10790","10791","10792","10793","10794","10795","10796","10797","10798","10799","10800","10801","10802","10803","10804","10805","10806","10807","10808","10809","10810","10811","10812","10813","10814","10815","10816","10817","10818","10819","10820","10821","10822","10823","10824","10825","10826","10827","10828","10829","10830","10831","10832","10833","10834","10835","10836","10837","10838","10839","10840","10841","10842","10843","10844","10845","10846","10847","10848","10849","10850","10851","10852","10853","10854","10855","10856","10857","10858","10859","10860","10861","10862","10863","10864","10865","10866","10867","10868","10869","10870","10871","10872","10873","10874","10875","10876","10877","10878","10879","10880","10881","10882","10883","10884","10885","10886","10887","10888","10889","10890","10891","10892","10893","10894","10895","10896","10897","10898","10899","10900","10901","10902","10903","10904","10905","10906","10907","10908","10909","10910","10911","10912","10913","10914","10915","10916","10917","10918","10919","10920","10921","10922","10923","10924","10925","10926","10927","10928","10929","10930","10931","10932","10933","10934","10935","10936","10937","10938","10939","10940","10941","10942","10943","10944","10945","10946","10947","10948","10949","10950","10951","10952","10953","10954","10955","10956","10957","10958","10959","10960","10961","10962","10963","10964","10965","10966","10967","10968","10969","10970","10971","10972","10973","10974","10975","10976","10977","10978","10979","10980","10981","10982","10983","10984","10985","10986","10987","10988","10989","10990","10991","10992","10993","10994","10995","10996","10997","10998","10999","11000","11001","11002","11003","11004","11005","11006","11007","11008","11009","11010","11011","11012","11013","11014","11015","11016","11017","11018","11019","11020","11021","11022","11023","11024","11025","11026","11027","11028","11029","11030","11031","11032","11033","11034","11035","11036","11037","11038","11039","11040","11041","11042","11043","11044","11045","11046","11047","11048","11049","11050","11051","11052","11053","11054","11055","11056","11057","11058","11059","11060","11061","11062","11063","11064","11065","11066","11067","11068","11069","11070","11071","11072","11073","11074","11075","11076","11077","11078","11079","11080","11081","11082","11083","11084","11085","11086","11087","11088","11089","11090","11091","11092","11093","11094","11095","11096","11097","11098","11099","11100","11101","11102","11103","11104","11105","11106","11107","11108","11109","11110","11111","11112","11113","11114","11115","11116","11117","11118","11119","11120","11121","11122","11123","11124","11125","11126","11127","11128","11129","11130","11131","11132","11133","11134","11135","11136","11137","11138","11139","11140","11141","11142","11143","11144","11145","11146","11147","11148","11149","11150","11151","11152","11153","11154","11155","11156","11157","11158","11159","11160","11161","11162","11163","11164","11165","11166","11167","11168","11169","11170","11171","11172","11173","11174","11175","11176","11177","11178","11179","11180","11181","11182","11183","11184","11185","11186","11187","11188","11189","11190","11191","11192","11193","11194","11195","11196","11197","11198","11199","11200","11201","11202","11203","11204","11205","11206","11207","11208","11209","11210","11211","11212","11213","11214","11215","11216","11217","11218","11219","11220","11221","11222","11223","11224","11225","11226","11227","11228","11229","11230","11231","11232","11233","11234","11235","11236","11237","11238","11239","11240","11241","11242","11243","11244","11245","11246","11247","11248","11249","11250","11251","11252","11253","11254","11255","11256","11257","11258","11259","11260","11261","11262","11263","11264","11265","11266","11267","11268","11269","11270","11271","11272","11273","11274","11275","11276","11277","11278","11279","11280","11281","11282","11283","11284","11285","11286","11287","11288","11289","11290","11291","11292","11293","11294","11295","11296","11297","11298","11299","11300","11301","11302","11303","11304","11305","11306","11307","11308","11309","11310","11311","11312","11313","11314","11315","11316","11317","11318","11319","11320","11321","11322","11323","11324","11325","11326","11327","11328","11329","11330","11331","11332","11333","11334","11335","11336","11337","11338","11339","11340","11341","11342","11343","11344","11345","11346","11347","11348","11349","11350","11351","11352","11353","11354","11355","11356","11357","11358","11359","11360","11361","11362","11363","11364","11365","11366","11367","11368","11369","11370","11371","11372","11373","11374","11375","11376","11377","11378","11379","11380","11381","11382","11383","11384","11385","11386","11387","11388","11389","11390","11391","11392","11393","11394","11395","11396","11397","11398","11399","11400","11401","11402","11403","11404","11405","11406","11407","11408","11409","11410","11411","11412","11413","11414","11415","11416","11417","11418","11419","11420","11421","11422","11423","11424","11425","11426","11427","11428","11429","11430","11431","11432","11433","11434","11435","11436","11437","11438","11439","11440","11441","11442","11443","11444","11445","11446","11447","11448","11449","11450","11451","11452","11453","11454","11455","11456","11457","11458","11459","11460","11461","11462","11463","11464","11465","11466","11467","11468","11469","11470","11471","11472","11473","11474","11475","11476","11477","11478","11479","11480","11481","11482","11483","11484","11485","11486","11487","11488","11489","11490","11491","11492","11493","11494","11495","11496","11497","11498","11499","11500","11501","11502","11503","11504","11505","11506","11507","11508","11509","11510","11511","11512","11513","11514","11515","11516","11517","11518","11519","11520","11521","11522","11523","11524","11525","11526","11527","11528","11529","11530","11531","11532","11533","11534","11535","11536","11537","11538","11539","11540","11541","11542","11543","11544","11545","11546","11547","11548","11549","11550","11551","11552","11553","11554","11555","11556","11557","11558","11559","11560","11561","11562","11563","11564","11565","11566","11567","11568","11569","11570","11571","11572","11573","11574","11575","11576","11577","11578","11579","11580","11581","11582","11583","11584","11585","11586","11587","11588","11589","11590","11591","11592","11593","11594","11595","11596","11597","11598","11599","11600","11601","11602","11603","11604","11605","11606","11607","11608","11609","11610","11611","11612","11613","11614","11615","11616","11617","11618","11619","11620","11621","11622","11623","11624","11625","11626","11627","11628","11629","11630","11631","11632","11633","11634","11635","11636","11637","11638","11639","11640","11641","11642","11643","11644","11645","11646","11647","11648","11649","11650","11651","11652","11653","11654","11655","11656","11657","11658","11659","11660","11661","11662","11663","11664","11665","11666","11667","11668","11669","11670","11671","11672","11673","11674","11675","11676","11677","11678","11679","11680","11681","11682","11683","11684","11685","11686","11687","11688","11689","11690","11691","11692","11693","11694","11695","11696","11697","11698","11699","11700","11701","11702","11703","11704","11705","11706","11707","11708","11709","11710","11711","11712","11713","11714","11715","11716","11717","11718","11719","11720","11721","11722","11723","11724","11725","11726","11727","11728","11729","11730","11731","11732","11733","11734","11735","11736","11737","11738","11739","11740","11741","11742","11743","11744","11745","11746","11747","11748","11749","11750","11751","11752","11753","11754","11755","11756","11757","11758","11759","11760","11761","11762","11763","11764","11765","11766","11767","11768","11769","11770","11771","11772","11773","11774","11775","11776","11777","11778","11779","11780","11781","11782","11783","11784","11785","11786","11787","11788","11789","11790","11791","11792","11793","11794","11795","11796","11797","11798","11799","11800","11801","11802","11803","11804","11805","11806","11807","11808","11809","11810","11811","11812","11813","11814","11815","11816","11817","11818","11819","11820","11821","11822","11823","11824","11825","11826","11827","11828","11829","11830","11831","11832","11833","11834","11835","11836","11837","11838","11839","11840","11841","11842","11843","11844","11845","11846","11847","11848","11849","11850","11851","11852","11853","11854","11855","11856","11857","11858","11859","11860","11861","11862","11863","11864","11865","11866","11867","11868","11869","11870","11871","11872","11873","11874","11875","11876","11877","11878","11879","11880","11881","11882","11883","11884","11885","11886","11887","11888","11889","11890","11891","11892","11893","11894","11895","11896","11897","11898","11899","11900","11901","11902","11903","11904","11905","11906","11907","11908","11909","11910","11911","11912","11913","11914","11915","11916","11917","11918","11919","11920","11921","11922","11923","11924","11925","11926","11927","11928","11929","11930","11931","11932","11933","11934","11935","11936","11937","11938","11939","11940","11941","11942","11943","11944","11945","11946","11947","11948","11949","11950","11951","11952","11953","11954","11955","11956","11957","11958","11959","11960","11961","11962","11963","11964","11965","11966","11967","11968","11969","11970","11971","11972","11973","11974","11975","11976","11977","11978","11979","11980","11981","11982","11983","11984","11985","11986","11987","11988","11989","11990","11991","11992","11993","11994","11995","11996","11997","11998","11999","12000","12001","12002","12003","12004","12005","12006","12007","12008","12009","12010","12011","12012","12013","12014","12015","12016","12017","12018","12019","12020","12021","12022","12023","12024","12025","12026","12027","12028","12029","12030","12031","12032","12033","12034","12035","12036","12037","12038","12039","12040","12041","12042","12043","12044","12045","12046","12047","12048","12049","12050","12051","12052","12053","12054","12055","12056","12057","12058","12059","12060","12061","12062","12063","12064","12065","12066","12067","12068","12069","12070","12071","12072","12073","12074","12075","12076","12077","12078","12079","12080","12081","12082","12083","12084","12085","12086","12087","12088","12089","12090","12091","12092","12093","12094","12095","12096","12097","12098","12099","12100","12101","12102","12103","12104","12105","12106","12107","12108","12109","12110","12111","12112","12113","12114","12115","12116","12117","12118","12119","12120","12121","12122","12123","12124","12125","12126","12127","12128","12129","12130","12131","12132","12133","12134","12135","12136","12137","12138","12139","12140","12141","12142","12143","12144","12145","12146","12147","12148","12149","12150","12151","12152","12153","12154","12155","12156","12157","12158","12159","12160","12161","12162","12163","12164","12165","12166","12167","12168","12169","12170","12171","12172","12173","12174","12175","12176","12177","12178","12179","12180","12181","12182","12183","12184","12185","12186","12187","12188","12189","12190","12191","12192","12193","12194","12195","12196","12197","12198","12199","12200","12201","12202","12203","12204","12205","12206","12207","12208","12209","12210","12211","12212","12213","12214","12215","12216","12217","12218","12219","12220","12221","12222","12223","12224","12225","12226","12227","12228","12229","12230","12231","12232","12233","12234","12235","12236","12237","12238","12239","12240","12241","12242","12243","12244","12245","12246","12247","12248","12249","12250","12251","12252","12253","12254","12255","12256","12257","12258","12259","12260","12261","12262","12263","12264","12265","12266","12267","12268","12269","12270","12271","12272","12273","12274","12275","12276","12277","12278","12279","12280","12281","12282","12283","12284","12285","12286","12287","12288","12289","12290","12291","12292","12293","12294","12295","12296","12297","12298","12299","12300","12301","12302","12303","12304","12305","12306","12307","12308","12309","12310","12311","12312","12313","12314","12315","12316","12317","12318","12319","12320","12321","12322","12323","12324","12325","12326","12327","12328","12329","12330","12331","12332","12333","12334","12335","12336","12337","12338","12339","12340","12341","12342","12343","12344","12345","12346","12347","12348","12349","12350","12351","12352","12353","12354","12355","12356","12357","12358","12359","12360","12361","12362","12363","12364","12365","12366","12367","12368","12369","12370","12371","12372","12373","12374","12375","12376","12377","12378","12379","12380","12381","12382","12383","12384","12385","12386","12387","12388","12389","12390","12391","12392","12393","12394","12395","12396","12397","12398","12399","12400","12401","12402","12403","12404","12405","12406","12407","12408","12409","12410","12411","12412","12413","12414","12415","12416","12417","12418","12419","12420","12421","12422","12423","12424","12425","12426","12427","12428","12429","12430","12431","12432","12433","12434","12435","12436","12437","12438","12439","12440","12441","12442","12443","12444","12445","12446","12447","12448","12449","12450","12451","12452","12453","12454","12455","12456","12457","12458","12459","12460","12461","12462","12463","12464","12465","12466","12467","12468","12469","12470","12471","12472","12473","12474","12475","12476","12477","12478","12479","12480","12481","12482","12483","12484","12485","12486","12487","12488","12489","12490","12491","12492","12493","12494","12495","12496","12497","12498","12499","12500","12501","12502","12503","12504","12505","12506","12507","12508","12509","12510","12511","12512","12513","12514","12515","12516","12517","12518","12519","12520","12521","12522","12523","12524","12525","12526","12527","12528","12529","12530","12531","12532","12533","12534","12535","12536","12537","12538","12539","12540","12541","12542","12543","12544","12545","12546","12547","12548","12549","12550","12551","12552","12553","12554","12555","12556","12557","12558","12559","12560","12561","12562","12563","12564","12565","12566","12567","12568","12569","12570","12571","12572","12573","12574","12575","12576","12577","12578","12579","12580","12581","12582","12583","12584","12585","12586","12587","12588","12589","12590","12591","12592","12593","12594","12595","12596","12597","12598","12599","12600","12601","12602","12603","12604","12605","12606","12607","12608","12609","12610","12611","12612","12613","12614","12615","12616","12617","12618","12619","12620","12621","12622","12623","12624","12625","12626","12627","12628","12629","12630","12631","12632","12633","12634","12635","12636","12637","12638","12639","12640","12641","12642","12643","12644","12645","12646","12647","12648","12649","12650","12651","12652","12653","12654","12655","12656","12657","12658","12659","12660","12661","12662","12663","12664","12665","12666","12667","12668","12669","12670","12671","12672","12673","12674","12675","12676","12677","12678","12679","12680","12681","12682","12683","12684","12685","12686","12687","12688","12689","12690","12691","12692","12693","12694","12695","12696","12697","12698","12699","12700","12701","12702","12703","12704","12705","12706","12707","12708","12709","12710","12711","12712","12713","12714","12715","12716","12717","12718","12719","12720","12721","12722","12723","12724","12725","12726","12727","12728","12729","12730","12731","12732","12733","12734","12735","12736","12737","12738","12739","12740","12741","12742","12743","12744","12745","12746","12747","12748","12749","12750","12751","12752","12753","12754","12755","12756","12757","12758","12759","12760","12761","12762","12763","12764","12765","12766","12767","12768","12769","12770","12771","12772","12773","12774","12775","12776","12777","12778","12779","12780","12781","12782","12783","12784","12785","12786","12787","12788","12789","12790","12791","12792","12793","12794","12795","12796","12797","12798","12799","12800","12801","12802","12803","12804","12805","12806","12807","12808","12809","12810","12811","12812","12813","12814","12815","12816","12817","12818","12819","12820","12821","12822","12823","12824","12825","12826","12827","12828","12829","12830","12831","12832","12833","12834","12835","12836","12837","12838","12839","12840","12841","12842","12843","12844","12845","12846","12847","12848","12849","12850","12851","12852","12853","12854","12855","12856","12857","12858","12859","12860","12861","12862","12863","12864","12865","12866","12867","12868","12869","12870","12871","12872","12873","12874","12875","12876","12877","12878","12879","12880","12881","12882","12883","12884","12885","12886","12887","12888","12889","12890","12891","12892","12893","12894","12895","12896","12897","12898","12899","12900","12901","12902","12903","12904","12905","12906","12907","12908","12909","12910","12911","12912","12913","12914","12915","12916","12917","12918","12919","12920","12921","12922","12923","12924","12925","12926","12927","12928","12929","12930","12931","12932","12933","12934","12935","12936","12937","12938","12939","12940","12941","12942","12943","12944","12945","12946","12947","12948","12949","12950","12951","12952","12953","12954","12955","12956","12957","12958","12959","12960","12961","12962","12963","12964","12965","12966","12967","12968","12969","12970","12971","12972","12973","12974","12975","12976","12977","12978","12979","12980","12981","12982","12983","12984","12985","12986","12987","12988","12989","12990","12991","12992","12993","12994","12995","12996","12997","12998","12999","13000","13001","13002","13003","13004","13005","13006","13007","13008","13009","13010","13011","13012","13013","13014","13015","13016","13017","13018","13019","13020","13021","13022","13023","13024","13025","13026","13027","13028","13029","13030","13031","13032","13033","13034","13035","13036","13037","13038","13039","13040","13041","13042","13043","13044","13045","13046","13047","13048","13049","13050","13051","13052","13053","13054","13055","13056","13057","13058","13059","13060","13061","13062","13063","13064","13065","13066","13067","13068","13069","13070","13071","13072","13073","13074","13075","13076","13077","13078","13079","13080","13081","13082","13083","13084","13085","13086","13087","13088","13089","13090","13091","13092","13093","13094","13095","13096","13097","13098","13099","13100","13101","13102","13103","13104","13105","13106","13107","13108","13109","13110","13111","13112","13113","13114","13115","13116","13117","13118","13119","13120","13121","13122","13123","13124","13125","13126","13127","13128","13129","13130","13131","13132","13133","13134","13135","13136","13137","13138","13139","13140","13141","13142","13143","13144","13145","13146","13147","13148","13149","13150","13151","13152","13153","13154","13155","13156","13157","13158","13159","13160","13161","13162","13163","13164","13165","13166","13167","13168","13169","13170","13171","13172","13173","13174","13175","13176","13177","13178","13179","13180","13181","13182","13183","13184","13185","13186","13187","13188","13189","13190","13191","13192","13193","13194","13195","13196","13197","13198","13199","13200","13201","13202","13203","13204","13205","13206","13207","13208","13209","13210","13211","13212","13213","13214","13215","13216","13217","13218","13219","13220","13221","13222","13223","13224","13225","13226","13227","13228","13229","13230","13231","13232","13233","13234","13235","13236","13237","13238","13239","13240","13241","13242","13243","13244","13245","13246","13247","13248","13249","13250","13251","13252","13253","13254","13255","13256","13257","13258","13259","13260","13261","13262","13263","13264","13265","13266","13267","13268","13269","13270","13271","13272","13273","13274","13275","13276","13277","13278","13279","13280","13281","13282","13283","13284","13285","13286","13287","13288","13289","13290","13291","13292","13293","13294","13295","13296","13297","13298","13299","13300","13301","13302","13303","13304","13305","13306","13307","13308","13309","13310","13311","13312","13313","13314","13315","13316","13317","13318","13319","13320","13321","13322","13323","13324","13325","13326","13327","13328","13329","13330","13331","13332","13333","13334","13335","13336","13337","13338","13339","13340","13341","13342","13343","13344","13345","13346","13347","13348","13349","13350","13351","13352","13353","13354","13355","13356","13357","13358","13359","13360","13361","13362","13363","13364","13365","13366","13367","13368","13369","13370","13371","13372","13373","13374","13375","13376","13377","13378","13379","13380","13381","13382","13383","13384","13385","13386","13387","13388","13389","13390","13391","13392","13393","13394","13395","13396","13397","13398","13399","13400","13401","13402","13403","13404","13405","13406","13407","13408","13409","13410","13411","13412","13413","13414","13415","13416","13417","13418","13419","13420","13421","13422","13423","13424","13425","13426","13427","13428","13429","13430","13431","13432","13433","13434","13435","13436","13437","13438","13439","13440","13441","13442","13443","13444","13445","13446","13447","13448","13449","13450","13451","13452","13453","13454","13455","13456","13457","13458","13459","13460","13461","13462","13463","13464","13465","13466","13467","13468","13469","13470","13471","13472","13473","13474","13475","13476","13477","13478","13479","13480","13481","13482","13483","13484","13485","13486","13487","13488","13489","13490","13491","13492","13493","13494","13495","13496","13497","13498","13499","13500","13501","13502","13503","13504","13505","13506","13507","13508","13509","13510","13511","13512","13513","13514","13515","13516","13517","13518","13519","13520","13521","13522","13523","13524","13525","13526","13527","13528","13529","13530","13531","13532","13533","13534","13535","13536","13537","13538","13539","13540","13541","13542","13543","13544","13545","13546","13547","13548","13549","13550","13551","13552","13553","13554","13555","13556","13557","13558","13559","13560","13561","13562","13563","13564","13565","13566","13567","13568","13569","13570","13571","13572","13573","13574","13575","13576","13577","13578","13579","13580","13581","13582","13583","13584","13585","13586","13587","13588","13589","13590","13591","13592","13593","13594","13595","13596","13597","13598","13599","13600","13601","13602","13603","13604","13605","13606","13607","13608","13609","13610","13611","13612","13613","13614","13615","13616","13617","13618","13619","13620","13621","13622","13623","13624","13625","13626","13627","13628","13629","13630","13631","13632","13633","13634","13635","13636","13637","13638","13639","13640","13641","13642","13643","13644","13645","13646","13647","13648","13649","13650","13651","13652","13653","13654","13655","13656","13657","13658","13659","13660","13661","13662","13663","13664","13665","13666","13667","13668","13669","13670","13671","13672","13673","13674","13675","13676","13677","13678","13679","13680","13681","13682","13683","13684","13685","13686","13687","13688","13689","13690","13691","13692","13693","13694","13695","13696","13697","13698","13699","13700","13701","13702","13703","13704","13705","13706","13707","13708","13709","13710","13711","13712","13713","13714","13715","13716","13717","13718","13719","13720","13721","13722","13723","13724","13725","13726","13727","13728","13729","13730","13731","13732","13733","13734","13735","13736","13737","13738","13739","13740","13741","13742","13743","13744","13745","13746","13747","13748","13749","13750","13751","13752","13753","13754","13755","13756","13757","13758","13759","13760","13761","13762","13763","13764","13765","13766","13767","13768","13769","13770","13771","13772","13773","13774","13775","13776","13777","13778","13779","13780","13781","13782","13783","13784","13785","13786","13787","13788","13789","13790","13791","13792","13793","13794","13795","13796","13797","13798","13799","13800","13801","13802","13803","13804","13805","13806","13807","13808","13809","13810","13811","13812","13813","13814","13815","13816","13817","13818","13819","13820","13821","13822","13823","13824","13825","13826","13827","13828","13829","13830","13831","13832","13833","13834","13835","13836","13837","13838","13839","13840","13841","13842","13843","13844","13845","13846","13847","13848","13849","13850","13851","13852","13853","13854","13855","13856","13857","13858","13859","13860","13861","13862","13863","13864","13865","13866","13867","13868","13869","13870","13871","13872","13873","13874","13875","13876","13877","13878","13879","13880","13881","13882","13883","13884","13885","13886","13887","13888","13889","13890","13891","13892","13893","13894","13895","13896","13897","13898","13899","13900","13901","13902","13903","13904","13905","13906","13907","13908","13909","13910","13911","13912","13913","13914","13915","13916","13917","13918","13919","13920","13921","13922","13923","13924","13925","13926","13927","13928","13929","13930","13931","13932","13933","13934","13935","13936","13937","13938","13939","13940","13941","13942","13943","13944","13945","13946","13947","13948","13949","13950","13951","13952","13953","13954","13955","13956","13957","13958","13959","13960","13961","13962","13963","13964","13965","13966","13967","13968","13969","13970","13971","13972","13973","13974","13975","13976","13977","13978","13979","13980","13981","13982","13983","13984","13985","13986","13987","13988","13989","13990","13991","13992","13993","13994","13995","13996","13997","13998","13999","14000","14001","14002","14003","14004","14005","14006","14007","14008","14009","14010","14011","14012","14013","14014","14015","14016","14017","14018","14019","14020","14021","14022","14023","14024","14025","14026","14027","14028","14029","14030","14031","14032","14033","14034","14035","14036","14037","14038","14039","14040","14041","14042","14043","14044","14045","14046","14047","14048","14049","14050","14051","14052","14053","14054","14055","14056","14057","14058","14059","14060","14061","14062","14063","14064","14065","14066","14067","14068","14069","14070","14071","14072","14073","14074","14075","14076","14077","14078","14079","14080","14081","14082","14083","14084","14085","14086","14087","14088","14089","14090","14091","14092","14093","14094","14095","14096","14097","14098","14099","14100","14101","14102","14103","14104","14105","14106","14107","14108","14109","14110","14111","14112","14113","14114","14115","14116","14117","14118","14119","14120","14121","14122","14123","14124","14125","14126","14127","14128","14129","14130","14131","14132","14133","14134","14135","14136","14137","14138","14139","14140","14141","14142","14143","14144","14145","14146","14147","14148","14149","14150","14151","14152","14153","14154","14155","14156","14157","14158","14159","14160","14161","14162","14163","14164","14165","14166","14167","14168","14169","14170","14171","14172","14173","14174","14175","14176","14177","14178","14179","14180","14181","14182","14183","14184","14185","14186","14187","14188","14189","14190","14191","14192","14193","14194","14195","14196","14197","14198","14199","14200","14201","14202","14203","14204","14205","14206","14207","14208","14209","14210","14211","14212","14213","14214","14215","14216","14217","14218","14219","14220","14221","14222","14223","14224","14225","14226","14227","14228","14229","14230","14231","14232","14233","14234","14235","14236","14237","14238","14239","14240","14241","14242","14243","14244","14245","14246","14247","14248","14249","14250","14251","14252","14253","14254","14255","14256","14257","14258","14259","14260","14261","14262","14263","14264","14265","14266","14267","14268","14269","14270","14271","14272","14273","14274","14275","14276","14277","14278","14279","14280","14281","14282","14283","14284","14285","14286","14287","14288","14289","14290","14291","14292","14293","14294","14295","14296","14297","14298","14299","14300","14301","14302","14303","14304","14305","14306","14307","14308","14309","14310","14311","14312","14313","14314","14315","14316","14317","14318","14319","14320","14321","14322","14323","14324","14325","14326","14327","14328","14329","14330","14331","14332","14333","14334","14335","14336","14337","14338","14339","14340","14341","14342","14343","14344","14345","14346","14347","14348","14349","14350","14351","14352","14353","14354","14355","14356","14357","14358","14359","14360","14361","14362","14363","14364","14365","14366","14367","14368","14369","14370","14371","14372","14373","14374","14375","14376","14377","14378","14379","14380","14381","14382","14383","14384","14385","14386","14387","14388","14389","14390","14391","14392","14393","14394","14395","14396","14397","14398","14399","14400","14401","14402","14403","14404","14405","14406","14407","14408","14409","14410","14411","14412","14413","14414","14415","14416","14417","14418","14419","14420","14421","14422","14423","14424","14425","14426","14427","14428","14429","14430","14431","14432","14433","14434","14435","14436","14437","14438","14439","14440","14441","14442","14443","14444","14445","14446","14447","14448","14449","14450","14451","14452","14453","14454","14455","14456","14457","14458","14459","14460","14461","14462","14463","14464","14465","14466","14467","14468","14469","14470","14471","14472","14473","14474","14475","14476","14477","14478","14479","14480","14481","14482","14483","14484","14485","14486","14487","14488","14489","14490","14491","14492","14493","14494","14495","14496","14497","14498","14499","14500","14501","14502","14503","14504","14505","14506","14507","14508","14509","14510","14511","14512","14513","14514","14515","14516","14517","14518","14519","14520","14521","14522","14523","14524","14525","14526","14527","14528","14529","14530","14531","14532","14533","14534","14535","14536","14537","14538","14539","14540","14541","14542","14543","14544","14545","14546","14547","14548","14549","14550","14551","14552","14553","14554","14555","14556","14557","14558","14559","14560","14561","14562","14563","14564","14565","14566","14567","14568","14569","14570","14571","14572","14573","14574","14575","14576","14577","14578","14579","14580","14581","14582","14583","14584","14585","14586","14587","14588","14589","14590","14591","14592","14593","14594","14595","14596","14597","14598","14599","14600","14601","14602","14603","14604","14605","14606","14607","14608","14609","14610","14611","14612","14613","14614","14615","14616","14617","14618","14619","14620","14621","14622","14623","14624","14625","14626","14627","14628","14629","14630","14631","14632","14633","14634","14635","14636","14637","14638","14639","14640","14641","14642","14643","14644","14645","14646","14647","14648","14649","14650","14651","14652","14653","14654","14655","14656","14657","14658","14659","14660","14661","14662","14663","14664","14665","14666","14667","14668","14669","14670","14671","14672","14673","14674","14675","14676","14677","14678","14679","14680","14681","14682","14683","14684","14685","14686","14687","14688","14689","14690","14691","14692","14693","14694","14695","14696","14697","14698","14699","14700","14701","14702","14703","14704","14705","14706","14707","14708","14709","14710","14711","14712","14713","14714","14715","14716","14717","14718","14719","14720","14721","14722","14723","14724","14725","14726","14727","14728","14729","14730","14731","14732","14733","14734","14735","14736","14737","14738","14739","14740","14741","14742","14743","14744","14745","14746","14747","14748","14749","14750","14751","14752","14753","14754","14755","14756","14757","14758","14759","14760","14761","14762","14763","14764","14765","14766","14767","14768","14769","14770","14771","14772","14773","14774","14775","14776","14777","14778","14779","14780","14781","14782","14783","14784","14785","14786","14787","14788","14789","14790","14791","14792","14793","14794","14795","14796","14797","14798","14799","14800","14801","14802","14803","14804","14805","14806","14807","14808","14809","14810","14811","14812","14813","14814","14815","14816","14817","14818","14819","14820","14821","14822","14823","14824","14825","14826","14827","14828","14829","14830","14831","14832","14833","14834","14835","14836","14837","14838","14839","14840","14841","14842","14843","14844","14845","14846","14847","14848","14849","14850","14851","14852","14853","14854","14855","14856","14857","14858","14859","14860","14861","14862","14863","14864","14865","14866","14867","14868","14869","14870","14871","14872","14873","14874","14875","14876","14877","14878","14879","14880","14881","14882","14883","14884","14885","14886","14887","14888","14889","14890","14891","14892","14893","14894","14895","14896","14897","14898","14899","14900","14901","14902","14903","14904","14905","14906","14907","14908","14909","14910","14911","14912","14913","14914","14915","14916","14917","14918","14919","14920","14921","14922","14923","14924","14925","14926","14927","14928","14929","14930","14931","14932","14933","14934","14935","14936","14937","14938","14939","14940","14941","14942","14943","14944","14945","14946","14947","14948","14949","14950","14951","14952","14953","14954","14955","14956","14957","14958","14959","14960","14961","14962","14963","14964","14965","14966","14967","14968","14969","14970","14971","14972","14973","14974","14975","14976","14977","14978","14979","14980","14981","14982","14983","14984","14985","14986","14987","14988","14989","14990","14991","14992","14993","14994","14995","14996","14997","14998","14999","15000","15001","15002","15003","15004","15005","15006","15007","15008","15009","15010","15011","15012","15013","15014","15015","15016","15017","15018","15019","15020","15021","15022","15023","15024","15025","15026","15027","15028","15029","15030","15031","15032","15033","15034","15035","15036","15037","15038","15039","15040","15041","15042","15043","15044","15045","15046","15047","15048","15049","15050","15051","15052","15053","15054","15055","15056","15057","15058","15059","15060","15061","15062","15063","15064","15065","15066","15067","15068","15069","15070","15071","15072","15073","15074","15075","15076","15077","15078","15079","15080","15081","15082","15083","15084","15085","15086","15087","15088","15089","15090","15091","15092","15093","15094","15095","15096","15097","15098","15099","15100","15101","15102","15103","15104","15105","15106","15107","15108","15109","15110","15111","15112","15113","15114","15115","15116","15117","15118","15119","15120","15121","15122","15123","15124","15125","15126","15127","15128","15129","15130","15131","15132","15133","15134","15135","15136","15137","15138","15139","15140","15141","15142","15143","15144","15145","15146","15147","15148","15149","15150","15151","15152","15153","15154","15155","15156","15157","15158","15159","15160","15161","15162","15163","15164","15165","15166","15167","15168","15169","15170","15171","15172","15173","15174","15175","15176","15177","15178","15179","15180","15181","15182","15183","15184","15185","15186","15187","15188","15189","15190","15191","15192","15193","15194","15195","15196","15197","15198","15199","15200","15201","15202","15203","15204","15205","15206","15207","15208","15209","15210","15211","15212","15213","15214","15215","15216","15217","15218","15219","15220","15221","15222","15223","15224","15225","15226","15227","15228","15229","15230","15231","15232","15233","15234","15235","15236","15237","15238","15239","15240","15241","15242","15243","15244","15245","15246","15247","15248","15249","15250","15251","15252","15253","15254","15255","15256","15257","15258","15259","15260","15261","15262","15263","15264","15265","15266","15267","15268","15269","15270","15271","15272","15273","15274","15275","15276","15277","15278","15279","15280","15281","15282","15283","15284","15285","15286","15287","15288","15289","15290","15291","15292","15293","15294","15295","15296","15297","15298","15299","15300","15301","15302","15303","15304","15305","15306","15307","15308","15309","15310","15311","15312","15313","15314","15315","15316","15317","15318","15319","15320","15321","15322","15323","15324","15325","15326","15327","15328","15329","15330","15331","15332","15333","15334","15335","15336","15337","15338","15339","15340","15341","15342","15343","15344","15345","15346","15347","15348","15349","15350","15351","15352","15353","15354","15355","15356","15357","15358","15359","15360","15361","15362","15363","15364","15365","15366","15367","15368","15369","15370","15371","15372","15373","15374","15375","15376","15377","15378","15379","15380","15381","15382","15383","15384","15385","15386","15387","15388","15389","15390","15391","15392","15393","15394","15395","15396","15397","15398","15399","15400","15401","15402","15403","15404","15405","15406","15407","15408","15409","15410","15411","15412","15413","15414","15415","15416","15417","15418","15419","15420","15421","15422","15423","15424","15425","15426","15427","15428","15429","15430","15431","15432","15433","15434","15435","15436","15437","15438","15439","15440","15441","15442","15443","15444","15445","15446","15447","15448","15449","15450","15451","15452","15453","15454","15455","15456","15457","15458","15459","15460","15461","15462","15463","15464","15465","15466","15467","15468","15469","15470","15471","15472","15473","15474","15475","15476","15477","15478","15479","15480","15481","15482","15483","15484","15485","15486","15487","15488","15489","15490","15491","15492","15493","15494","15495","15496","15497","15498","15499","15500","15501","15502","15503","15504","15505","15506","15507","15508","15509","15510","15511","15512","15513","15514","15515","15516","15517","15518","15519","15520","15521","15522","15523","15524","15525","15526","15527","15528","15529","15530","15531","15532","15533","15534","15535","15536","15537","15538","15539","15540","15541","15542","15543","15544","15545","15546","15547","15548","15549","15550","15551","15552","15553","15554","15555","15556","15557","15558","15559","15560","15561","15562","15563","15564","15565","15566","15567","15568","15569","15570","15571","15572","15573","15574","15575","15576","15577","15578","15579","15580","15581","15582","15583","15584","15585","15586","15587","15588","15589","15590","15591","15592","15593","15594","15595","15596","15597","15598","15599","15600","15601","15602","15603","15604","15605","15606","15607","15608","15609","15610","15611","15612","15613","15614","15615","15616","15617","15618","15619","15620","15621","15622","15623","15624","15625","15626","15627","15628","15629","15630","15631","15632","15633","15634","15635","15636","15637","15638","15639","15640","15641","15642","15643","15644","15645","15646","15647","15648","15649","15650","15651","15652","15653","15654","15655","15656","15657","15658","15659","15660","15661","15662","15663","15664","15665","15666","15667","15668","15669","15670","15671","15672","15673","15674","15675","15676","15677","15678","15679","15680","15681","15682","15683","15684","15685","15686","15687","15688","15689","15690","15691","15692","15693","15694","15695","15696","15697","15698","15699","15700","15701","15702","15703","15704","15705","15706","15707","15708","15709","15710","15711","15712","15713","15714","15715","15716","15717","15718","15719","15720","15721","15722","15723","15724","15725","15726","15727","15728","15729","15730","15731","15732","15733","15734","15735","15736","15737","15738","15739","15740","15741","15742","15743","15744","15745","15746","15747","15748","15749","15750","15751","15752","15753","15754","15755","15756","15757","15758","15759","15760","15761","15762","15763","15764","15765","15766","15767","15768","15769","15770","15771","15772","15773","15774","15775","15776","15777","15778","15779","15780","15781","15782","15783","15784","15785","15786","15787","15788","15789","15790","15791","15792","15793","15794","15795","15796","15797","15798","15799","15800","15801","15802","15803","15804","15805","15806","15807","15808","15809","15810","15811","15812","15813","15814","15815","15816","15817","15818","15819","15820","15821","15822","15823","15824","15825","15826","15827","15828","15829","15830","15831","15832","15833","15834","15835","15836","15837","15838","15839","15840","15841","15842","15843","15844","15845","15846","15847","15848","15849","15850","15851","15852","15853","15854","15855","15856","15857","15858","15859","15860","15861","15862","15863","15864","15865","15866","15867","15868","15869","15870","15871","15872","15873","15874","15875","15876","15877","15878","15879","15880","15881","15882","15883","15884","15885","15886","15887","15888","15889","15890","15891","15892","15893","15894","15895","15896","15897","15898","15899","15900","15901","15902","15903","15904","15905","15906","15907","15908","15909","15910","15911","15912","15913","15914","15915","15916","15917","15918","15919","15920","15921","15922","15923","15924","15925","15926","15927","15928","15929","15930","15931","15932","15933","15934","15935","15936","15937","15938","15939","15940","15941","15942","15943","15944","15945","15946","15947","15948","15949","15950","15951","15952","15953","15954","15955","15956","15957","15958","15959","15960","15961","15962","15963","15964","15965","15966","15967","15968","15969","15970","15971","15972","15973","15974","15975","15976","15977","15978","15979"],["TSPAN6","DPM1","SCYL3","C1orf112","FGR","CFH","FUCA2","GCLC","NFYA","STPG1","NIPAL3","LAS1L","ENPP4","SEMA3F","CFTR","ANKIB1","CYP51A1","KRIT1","RAD52","BAD","LAP3","CD99","HS3ST1","AOC1","HECW1","MAD1L1","LASP1","SNX11","TMEM176A","M6PR","KLHL13","ICA1","DBNDD1","ALS2","CASP10","CFLAR","TFPI","NDUFAF7","RBM5","MTMR7","SLC7A2","ARF5","SARM1","POLDIP2","PLXND1","AK2","CD38","FKBP4","KDM1A","RBM6","CAMKK1","RECQL","VPS50","ARHGAP33","NDUFAB1","PDK4","SLC22A16","ZMYND10","ABCB5","ARX","SLC25A13","ST7","CDC27","SLC4A1","HCCS","DVL2","PRSS22","UPF1","SKAP2","SLC25A5","MCUB","POLR2J","DHX33","THSD7A","LIG3","RPAP3","ACSM3","REXO5","CIAPIN1","SPPL2B","FAM214B","COPZ2","PRKAR2B","MSL3","CREBBP","TSPOAP1","MPO","GCFC2","WDR54","CROT","ABCB4","KMT2E","RHBDD2","IBTK","ZNF195","MYCBP2","FBXL3","ITGAL","PDK2","ITGA3","ZFX","LAMP2","ITGA2B","ASB4","GDE1","REX1BD","CRLF1","OSBPL7","TMEM98","YBX2","MAP3K14","ABCC8","TMEM132A","AP2B1","ZNF263","CX3CL1","SPATA20","CACNA1G","TNFRSF12A","DLX6","MAP3K9","RALA","BAIAP2L1","KDM7A","ETV1","AGK","ALDH3B1","TTC22","PHTF2","FARP2","USH1C","GGCT","DBF4","TBXA2R","IFRD1","LGALS14","COX10","GTF2IRD1","PAF1","VPS41","ARHGAP44","ELAC2","SCIN","ARSD","PNPLA4","ADIPOR2","CDKL3","UPP2","MARK4","PROM1","CCDC124","CEACAM21","PAFAH1B1","NOS2","DNAH9","KIAA0100","SLC13A2","GAS7","TRAPPC6A","MATK","CEACAM7","ST7L","TKTL1","PAX6","RPUSD1","RHBDF1","LUC7L","CACNA2D2","BAIAP3","TSR3","PIGQ","CRAMP1","TEAD3","DNAJC11","FMO3","MYLIP","NOX1","E2F2","PSMB1","JARID2","CDKL5","CAMK1G","CDK11A","NADK","DLEC1","CYTH3","ADAM22","SYPL1","CYB561","SPAG9","CELSR3","AASS","PLEKHG6","SS18L2","MPND","MGST1","CRY1","PGLYRP1","NFIX","ST3GAL1","MMP25","IL32","PKD1","MAPK8IP2","MED24","RHOBTB2","HEATR5B","SEC62","RPS20","CSDE1","UBE3C","REV3L","TENM1","PAX7","MASP2","IYD","FAM76A","TRAF3IP3","POMT2","VTA1","MLXIPL","BAZ1B","RANBP9","ETV7","SPRTN","EEF1AKNMT","DYRK4","ZNF207","UQCRC1","STARD3NL","CD9","HHATL","NCAPD2","IFFO1","GIPR","PHF7","NISCH","STAB1","FUZ","SLC6A13","IDS","ZNF200","CD4","LRRC23","BTK","HFE","SCMH1","FYN","HIVEP2","FMO1","ELOA","LYPLA2","CLCN6","MRC2","TSPAN9","BTBD7","APBA3","MKS1","ABHD5","ANOS1","AKAP8L","MBTD1","UTP18","RNF216","TTC19","PTBP1","DPF1","SYT7","LARS2","PIK3C2A","PLAUR","ANLN","WIZ","GPR21","DCN","QPCTL","PPP5C","CEP68","MAP4K3","ZBTB32","TYROBP","TMEM159","BRCA1","ERCC1","CD22","SEMA3B","MBTPS2","PRICKLE3","LTF","EXTL3","ELOVL5","ALOX5","KDM5D","CALCOCO1","UBR7","MAP4K5","EHD3","PSMC4","MAN2B2","SLC7A14","CLDN11","SLC25A39","MVP","NUB1","PGM3","RWDD2A","CLK1","POLR3B","ANGEL1","RNF14","DNASE1L1","DDX11","HEBP1","GPRC5A","MAMLD1","CD6","TACC3","UFL1","POLA2","ZC3H3","CAPN1","ACPP","SLC30A9","MTMR11","COX15","CCDC88C","YAF2","ZMYND11","WAS","BID","MATR3","XYLT2","RGPD5","STMN4","NUDCD3","ISL1","CHDH","IL20RA","CLCA4","GLT8D1","ATP2C1","IGF1","SLC38A5","RALBP1","RUFY3","CNTN1","SLC11A1","WWTR1","AGPS","CXorf56","TTC27","ZNF582","VSIG2","PHLDB1","CYP24A1","PRDM11","SYT13","SNAI2","CD74","HGF","ZRANB1","NCDN","ADGRA2","ZFP64","MNAT1","SAMD4A","RUNX3","MRE11","PLEKHB1","SERPINB1","CYP3A43","SLC7A9","SPAST","NRXN3","OSBPL5","AQR","CPS1","FHL1","RTF2","GABRA1","NLRP2","SLC45A4","RNF10","ZNF839","ZDHHC6","GRAMD1B","RNH1","NDUFS1","RB1CC1","ERP44","ALAS1","BIRC3","AKAP11","GLRX2","SNAPC1","DERA","STRAP","ABCC2","DEF6","PLEKHO1","GCLM","UBR2","EHD2","DEPDC1","CCDC28A","RRAGD","HSF2","PHF20","NR1H3","TYMP","NCAPH2","TOMM34","SEC63","KPNA6","VIM","RTEL1-TNFRSF6B","FAS","RNASET2","CD44","KCNG1","AGPAT4","SLAMF7","BTN3A1","MIPEP","PRKCH","IFNGR1","B4GALT7","SH2D2A","VRK2","TNFRSF1B","VEZT","POU2F2","BRD9","SNX1","TBPL1","ARNTL2","BCLAF1","SLC39A9","ANK1","TFB1M","RABEP1","HMGB3","NUP160","BAK1","MUSK","IKZF2","GRN","FAM13B","ARHGAP31","CENPQ","SARS1","RANBP3","ARID4A","EIPR1","PNPLA6","IFT88","ALG1","ZCCHC8","ABCF2","CHPF2","LRRC7","FUT8","UBA6","GAB2","ATP6V0A1","PIAS1","SLC4A7","APBA2","MAP2K3","EFCAB1","TMSB10","ASTE1","RNF19A","PEX3","GABARAPL2","SH3YL1","FAM136A","VCL","DEPDC1B","DAPK2","NSMAF","ADSS2","STAP1","TIMP2","RFC1","TBC1D23","CUL3","MYOM2","CYP46A1","ZZZ3","SLC18A1","USP2","CASR","TUBG2","RPL26L1","FLT4","NSUN2","FBXO42","MFAP3","MRI1","METTL1","AGA","PI4K2B","BOD1L1","MAT2B","TLL1","EDC4","TRIO","VCAN","CLEC16A","MSR1","CDH1","MTREX","DNAH5","ZFYVE16","RIPOR1","RAI14","PNKP","BEST2","PHLPP2","SPDL1","STAU2","SLC66A1","CTNS","PHF23","INPP4A","RAB27B","PSMA4","MYO16","LSG1","PARP3","TNC","THAP3","RIPOR3","TDP1","AIFM2","C2orf83","SPATA7","MED17","RETSAT","CAPG","AP2S1","USH2A","TG","ADAM28","BARX2","DCUN1D1","JADE2","LCP2","TRIT1","ADRB1","CUL7","CTNNA1","PHKA2","CNTLN","EPHA3","HSPA5","DSG2","GEMIN8","OFD1","GPM6B","PREX2","WDR37","YTHDC2","CTPS2","ATP6V1H","POLR2B","FAM214A","ARAP2","TPR","CP","KIAA0556","DTNBP1","XK","ANO2","C12orf4","SCML1","WWC3","ARHGAP6","FAM184B","MAP4","GOPC","USP28","HDAC9","TSPAN17","NOP16","CC2D2A","RRM2B","ZNF800","SNX29","LMO3","MRPS10","RSF1","VPS13D","CELF2","FAM120A","R3HDM1","COL9A2","KITLG","ERCC8","ADAMTS6","H6PD","VAMP3","PER3","TNFRSF9","EPN3","LTBP1","RCN1","ELN","RFC2","ARID1B","CLPTM1L","NEDD4L","FOXP3","PPP1R3F","HEXB","PTCD2","NEXMIF","JKAMP","DKK3","ARHGEF5","NFE2L3","MCUR1","LIMA1","LETMD1","SLC4A8","PTGER3","TNIP3","MAPK9","COL23A1","BCAR1","FAM160A2","HERPUD1","HOMER3","RAD51","POLQ","PIK3CB","CYBA","THOC3","HEBP2","MPHOSPH9","PLEKHA5","PRSS8","SIKE1","RRP12","FNIP2","MSMO1","TTC17","FSTL4","FOXN3","AKR7A2","MRTO4","NNAT","USE1","MCF2L2","NRIP2","LAMA3","AP5M1","ANAPC4","KCNQ1","TRAPPC3","THRAP3","PHPT1","ENTPD2","LY75","ARID4B","OPN3","SDCCAG8","HHAT","KIF1B","FOXC1","TBC1D22A","SYNE2","PLEKHH1","ATP9A","FAM168A","RELT","GALC","NOP58","SZRD1","CUL1","FAM114A2","CYFIP2","TAB2","GINM1","EIF2AK2","USP36","KMT2C","MCOLN3","PUM2","MRPL43","ITIH4","ITIH1","HPF1","ZFR","ZNF280C","TRAF1","RC3H2","IL17RB","TRAF3IP2","GYG2","DCBLD2","SERPINB3","SOAT1","PKP2","GDI2","PRDM1","ATG5","TMCC3","PITHD1","MTA3","USP13","ATP11B","LAMC2","CDK14","SEC61A1","PPP1R12A","RASGRF1","CAMK2B","CROCC","POLR3E","ATP2B4","ZC3H11A","RIOK2","YIPF1","NDC1","DGKG","FLYWCH1","UNKL","TBXAS1","PARP12","ALDH18A1","TARBP1","GATB","MXD1","CDK17","DNAJC25","SLC2A3","PSD","CTDP1","YBX3","STYK1","WNK1","CCAR1","OGFR","GNA15","PIGV","PTPRU","SNRNP40","RIMBP2","QSER1","MPC1","ACAA1","BCAT1","HDAC7","LZTS1","PRDM6","SPAG4","NCKAP1","MRPS35","GUCY1B1","SFSWAP","TNK2","MON2","CDH3","GPBP1","DGAT2","ZNF112","CS","LTK","MRPS24","ELMO2","WAPL","VMP1","APPBP2","POLD1","SEZ6","EIF4B","SLC6A16","BICRA","SPHK2","RPL18","CA11","ISOC2","U2AF2","EPN1","MED29","ZNF275","MTMR1","GPC1","ADCK1","HAGH","RNF4","CASP8","LIMCH1","INTS13","TM7SF3","SPA17","TSPAN32","CCN5","DMRT3","ST3GAL6","ATP2C2","NGFR","CDON","TAF2","HIPK2","TNPO3",null,"RFXANK","TMEM161A","LPAR2","CTSA","SUGP2","SLC12A2","SNX24","EYA2","CNN2","ABCA7","SNCAIP","DDX20","BTBD1","FAR2","BCAS1","CHI3L2","SBNO2","PMS1","HMG20B","CALCRL","TAF11","ANKS1A","AP3D1","ZNF76","SLC9A3R2","NTHL1","UHRF1BP1","GNAI3","IPO5","OAT","WDR3","PKN2","WDR18","TRAM2","NTN1","MCM10","DGKA","ERBB3","ANKRD44","KARS1","ADAT1","PDIA5","TBC1D22B","NDUFB4","SPEN","MYLK","ZC3H15","MAP2K4","PACC1","SLK","CYB5R4","COL17A1","GSTO2","SEC61A2","PRKCQ","TLE2","ASB1","FAM107B","ME1","TBC1D1","CDK13","MTHFD2","SLC9A7","FOXJ2","YBX1","PDE4A","PPP2R5A","CTNNA2","ELAVL1","TIE1","DIP2B","SMARCD1","KDM4A","NFYC","ZMYND12","SLC9A3","NGEF","ASPM","CD84","ELOVL1","SPI1","ZNRD1","MPPED2","CLDN18","ZBTB11","ATXN3","GOLGA5","FGFR2","LRRC40","ISOC1","EML1","TRMT11","THUMPD1","MSANTD3","ATG2B","ARFGEF1","ACSM2B","ZFAT","MTFR1","STAG3","FECH","MYO9A","DDX3Y","PFKP","IDI1","SP100","KLF6","PLPP1","NEO1","TRAM1","PHKA1","TNFRSF1A","CACNB1","EVI5","STOML1","PKM","DHX29","DNTTIP2","METTL22","TP53BP1","TRO","RRP15","RHOA","DHX8","PRKCZ","ZFY","IARS2","NAV3","IDH3G","ROGDI","PDZD4","ROCK1","CBFB","PDK3","HYAL2","HDAC4","RASSF1","FGFR3","IFI35","HEATR6","COASY","PLEKHH3","MEF2A","OTUD5","TFE3","TBC1D25","ACSL4","INPP5A","GPKOW","GRIPAP1","FTSJ1","PRR11","REEP1","ATP11A","POLR1A","LAPTM4A","TTC7A","IP6K2","SRBD1","KIF2A","RASGRP2","PSME4","IFT80","SIRT2","ERLEC1","PPP2R5B","PYGM","PITX1","MAST4","ADGRF5","SDK2","NUP133","NUCKS1","VPS35","DNAJA2","BCL3","KCNAB2","ABCC9","CLEC2D","FUNDC1","MAOB","RORA","DRD4","TGFBR3","PLA2G10","HES2","ATP1B3","NEDD4","PIGB","MAPK6","GNB5","RAB27A","HDHD5","UFD1","LRP6","GUCY2C","PHRF1","ELP1","NUCB2","PFN2","PTPN3","SPTB","DAPP1","FGF10","SLC44A1","TMEM260","SMG6","EXOC5","CLTCL1","FSTL3","DGCR2","RNF126","MNT","ZXDC","JMJD6","POLB","ST6GALNAC1","WIPI1","FRMPD1","GBA2","NDST1","ASNS","AP3M2","CNGB1","ST6GALNAC2","CHAT","PABPC1","TESK2","CFAP20","CSNK2A2","PTPN21","EIF2B3","CAMK2A","TCOF1","CDC42","OSBPL3","SLC12A3","RAD18","ATP2B1","TRPM5","NCK2","MAP4K4","MGAT4A","RPL31","WDR1","SNX13","ARHGAP10","RPS6KA2","ING3","VASH1","LMCD1","BUD23","SEL1L","TRIP13","ATP6AP1","TCF3","TRIB2","DAZAP1","MBD3","HLTF","FAM50A","FAM3A","CPSF1","MYO3B","CYBRD1","CDH19","PDCD2","SLC6A15","RDH11","PRKACA","ADGRL1","SPP2","ACTN1","ZFYVE26","RPS6KA6","EPN2","PTPN18","LIMS2","ASIC4","SPEG","LNX1","ALDH3A2","TFRC","SREBF1","TRPC5","AFF4","UBE2D1","MPP5","RHOBTB1","SMC1A","HSD17B10","MARK2","HMMR","CHFR","TRHDE","P4HA2","FCGR2B","NFATC3","TRNT1","ACADVL","STK10","FBXW11","ACAP1","CRMP1","EVC","DERL2","SIDT1","NDE1","MRVI1","TMEM38A","AP1M1","PVR","XRCC1","SCARB1","CYP2W1","MCM2","PANX2","SELENOO","TP63","ALPK1","LLGL2","PDE8A","CLCN4","NLE1","SDHA","SMARCE1","GSDMB","KDM5A","PPP2R3A","FERMT2","ABCB11","DHRS9","PTGS2","IGF2BP2","MAP3K13","ST6GAL1","TBX21","FRY","PICALM","NSF","GLI2","CLASP1","MRPS34","NOTCH3","CLNS1A","PPP2R2C","EED","CDHR2","TSG101","NCBP3","ATP2A3","CA12","MGLL","NTN4","BCS1L","NUAK1","DPP8","SLC24A1","ZNF532","SCARF1","LMAN1","HACD3","IPCEF1","ZZEF1","ENO1","SLC12A1","MYDGF","ANO8","TUBE1","ARHGEF10L","TXK","TACR2","ACTR6","TIPIN","SRI","EIF4G3","NUP37","SEMA3A","GTSE1","SEMA3C","TTC38","ACAT1","GRAMD4","CELSR1","ZNF638","SLC25A40","TIMM21","ADD2","RASAL2","VPS9D1","ZNF37A","MARK3","SLC25A3","FNDC3B","FOSL2","CACNG5","CACNG4","FRYL","TMEM131","FSCN1","ACTB","MOCOS","PLD1","ATP12A","WDR62","DLG1","RAB7A","BCAP29","SEC31B","SART3","ARHGAP15","TUBA3D","EXOSC7","KIFAP3","MKRN2","MCM6","REXO2","RBM7","RBMS2","BAZ2A","PTPN23","MLH1","UNG","FMO4","KLHL20","RGS11","SLC46A1","PLXNA2","SPAG5","ANKRD13A","TPD52","ACACB","TRAF4","PAG1","GPATCH1","ICAM3","NT5C2","MCAM","GPC4","MBNL3","CAMSAP3","RAP1GAP","XAB2","ARHGEF1","STXBP2","MAP2K7","DGKD","CTTNBP2","RARB","TOP2B","TM9SF3","NFKB2","UBE2T","PPP1R12B","DNAJC10","GTF3C1","IL4R","USP33","PAK3","DCX","SNRPA","SPAG6","EXOSC5","DYNC1I2","APBB1IP","LRCH4","FAM76B","SIRT6","TYR","POLD3","CAPZB","GPR137B","NAALAD2","JADE1","SLC25A43","UBE2A","FGFR1","FKBP6","SMC1B","FBLN1","ITGA8","CST7","MAP2","PIAS2","AMPH","ARAF","MCCC1","LAMP3","FAP","NEBL","ACER3","UBE2K","PIK3C3","N4BP2","TIGAR","TULP3","SYNJ2","ADCY2","PPP2R5C","RBFOX1","GNB1","EDN1","MLLT10","ZCWPW1","P2RY10","ITM2A","NRDC","VDAC3","PCM1","TNRC6C","CBFA2T2","ITCH","PKD2L2","TP53INP2","SDF4","MYH7B","BPIFB2","TP73","TOLLIP","UBE2D4","CLUL1","RUNX1T1","CDH17","THOC1","FKBP7","OSBPL6","SLC1A3","XRCC5","LXN","SP140","MKNK1","TNS1","REXO1","SAR1A","CDC14A","RAPGEF3","CEACAM1","SENP1","CIC","LIPE","FDFT1","PAFAH1B3","OPHN1","KIF22","CARMIL1","PGM1","DDX1","DNM2","EPB41L2","RIMS1","STX7","RABL2B","KEAP1","DDX43","PTPRH","SLC35C2","CRYBG3","SCTR","RFX3","RIF1","RAB21","SLC4A4","SMARCA2","SESN1","MID2","PIH1D3","SRCAP","PUM3","CHRNA3","KCNN2","CNOT4","PSEN1","CPOX","CLDND1","MOK","HSP90AA1","RBL1","DLGAP4","IGSF9B","NDC80","AP4E1","RSBN1","MAGI3","CXCL2","COL4A4","TCF7","OSTM1","CDH7","IMPG2","PCNP","EXD2","ARG2","MEF2C","PTPRC","PKP1","UBA5","STK17B","CDC14B","ZNF510","LRP2","ZNF506","JMJD4","DUSP12","AACS","DELE1","CADPS2","PCDHB4","HSPB11","PHLPP1","ATP8B1","IL12RB2","SMARCD3","WDR70","FYB1","MPP4","STRADB","BZW1","C1QTNF3","ME2","C5orf22","CCNT2","FAM135A","COL19A1","EPB41L3","COBLL1","DLG3","KCNK2","SERTAD4","TRAF5","MRPL22","GEMIN5","NFE2L1","SEMA5B","GSK3B","ITGB5","ERC1","XPO1","RNF13","TRPM3","PALB2","DOP1A","LYRM2","BCKDHB","KAT6A","TUT7","ULK2","GRHL2","TNPO1","PLOD1","P2RX5","ITGAE","DIS3","PIBF1","TDRD3","NUFIP1","PDS5B","OXCT1","RRAGB","CYLD","SLC27A5","ZNF324","ZNF671","ZNF416","ZNF586","ZNF446","ZNF264","RPS5","FAT1","YTHDC1","CHMP2B","SMAP2","PPIE","ZMPSTE24","STARD7","NOA1","REST","HAL","SSH1","GSTP1","APLP2","FAM234B","SLCO1A2","WBP11","EIF3I","COL16A1","TXLNA","APOB","NCOA1","AGBL5","EFR3B","KIF3C","RAB10","GCKR","HADHA","MAPRE3","CAD","CD59","CD82","BCORL1","ATRX","AK6","FCN1","MYNN","MECOM","SCAMP1","PREP","HACE1","SEH1L","WDR47","WDFY1","OVGP1","SLC25A24","MAP3K4","PILRA","IGSF9","ABCB1","ZNF213","AKR1B1","CPNE3","RRN3","CTTN","MTIF2","DDHD2","TTC39A","EPS15","ORC1","MGST2","CHERP","ATG16L1","USP40","POMGNT1","RAD54L","MAST2","DNAJA1","B4GALT1","CHMP5","NFX1","AQP6","DIMT1","IPO11","FOLH1","EIF2AK1","EPDR1","SNX10","SEPHS1","MRPL28","ITPKC","CEACAM6","FAT2","RBM22","TMED2","ERO1B","ZFAND6","HSD17B2","TXLNG","LAT2","HUWE1","ZW10","ALG9","NOX4","ACOX3","MTMR2","PPP1R15A","TRIP6","ACHE","FTL","SRRT","BAX","NLK","PIGS","ADAMTS2","TMPRSS11E","ATXN7L3","PGS1","PSMC5","UIMC1","CETP","MT3","LPCAT2","GNAO1","OGFOD1","SH3BP2","NOP14","ADD1","L2HGDH","TXNDC16","RTRAF","NID2","GMCL1","SF3B2","KLHL42","GNAS","DNM1L","PHACTR3","ERGIC2","TFAP2C","AURKA","CASS4","PIR","AAMDC","RFX2","METTL2A","SULT2B1","ALG6","CNOT3","GP6","PTPN4","DDX18","KHSRP","GNA11","ASAP3","EDEM2","DNMT3B","REM1","TPX2","PDRG1","EPB41L1","SLC15A1","DOCK9","ANKRD10","TGDS","DOCK3","C3orf18","COQ9","TMEM40","KIF9","ARHGAP28","CRLS1","PPP1R13B","ATRN","SMOX","SIGLEC1","FKBP1A","NSFL1C","SLC4A11","C20orf194","ZNF343","EBF4","CPXM1","MAVS","LZTS3","XRN2","KIZ","DYNLL1","TESC","SNX5","RPL6","SIRPG","MAPKAPK5","P2RX7","ESF1","RBBP9","ANAPC5","SLC23A2","SLC8B1","TMEM230","DZANK1","KDM2B","CFAP61","TASP1","OAS1","GCN1","RPLP0","PXN","RPH3A","KIF16B","TRMT6","CHGB","PEBP1","BRAP","ERP29","NOS1","FUS","IGBP1","FXYD5","ZNF302","GRAMD1A","FXYD3","CDIP1","CMTM1","KCNH4","GANAB","GMIP","RBM41","BIRC5","LAG3","MLF2","OTUB2","DDX24","ZBTB25","NECAP1","ARHGAP4","ANKRD24","DHX32","RCOR1","GPATCH2L","LTBP4","BLVRB","SLC9A1","SPTLC1","PAPOLA","CCNK","PCBP4","RGS1","YPEL3","MRPS33","NDUFB2","NUDC","MAEA","ICAM1","STRN4","IRAK3","LYZ","MUL1","TFAP4","PDCD7","SPG21","DNAJB11","P3H2","CHRD","FLT3LG","RAB11FIP3","GNPTG","ZNF268","GOLGA3","PABPC4","CD209","CERS4","MCOLN1","USP48","EFNB1","PDPR","AARS1","GLG1","KIF4A","TNRC6A","PLEKHG2","NAT14","PITPNM2","EXOC1","RBM27","OSBPL8","DTX2","NLRC4","PUS7","NRCAM","LAMB1","SLC26A4","DLD","WDR7","TXNL1","IL5RA","ABCC6","CMTM6","ITGA6","RAPGEF4","MAP3K20","FH","SEL1L3","TF","CDV3","MYO15A","ALKBH5","NLRP1","PITPNM3","SPAG7","ORC6","ZFHX4","SLC17A6","ZC3HC1","ESR1","RGS17","ANGPT2","TMEM101","CD200","CCDC80","PSME1","PPP2R3C","HAUS4","SLC7A8","OSGEP","SLC22A17","RNF31","SCFD1","G2E3","HECTD1","HNRNPC","RPGRIP1","SUPT16H","TOX4","GEMIN2","TGM1","TINF2","SEMA6A","TRPM7","TYRO3","WDR76","CAPN3","SNAP23","PHGDH","COL9A3","EZR","MYL6","AGO1","TEKT2","CLSPN","RFFL","UNC13D","MFSD11","DPYSL2","TGFB2","GPATCH2","NUP50","COMT","ADA2","VNN3","ECHDC1","LRRFIP2","SEC22C","XYLB","HDAC6","OR1I1","GABRP","CDC6","UPRT","CDC23","AAAS","CBX5","FMO2","SUCO","MSH2","MAP3K1","DHPS","HOOK2","ARCN1","EPB41L4B","TMEM38B","PSMD5","PTGS1","NUP188","CRAT","SH2D3C","NANS","TBC1D2","WHRN","PDE6C","CWF19L1","SEMA4G","BTAF1","IKZF5","BLNK","TLL2","CYP26A1","TDRD1","SORBS1","BAMBI","WAC","CREM","NUBP2","HIVEP1","TREM2","CRISP3","FKBP5","SRPK1","BRPF3","MRPS18A","TMEM14A","EFHC1","HSP90AB1","CDC5L","ITPR3","ZNF184","DSP","SIRT1","HNRNPH3","IFT74","JAK2","IL12RB1","ABL1","ACOT7","SH3GLB1","CDC7","SYDE2","PCSK5","SCD","TMED1","ABLIM1","ERMP1","RAB18","NRP1","PALMD","TSPAN15","MACROH2A2","WASHC2A","MAST3","MZF1","OCEL1","MYO9B","KCNK6","CATSPERG","PSMD8","FBXL19","STX1B","HSD3B7","SETD1A","BCL7C","EFNA2","CIRBP","ATP5F1D","CBARP","PCDH11Y","PRKY","IGFALS","HNRNPM","MARCHF2","NDUFB7","TECR","TIMM13","CDC34","MTAP","MISP","CEP170B","POLR2E","POLRMT","CDHR5","IZUMO4","RASSF7","GADD45B","PALM","MADCAM1","MKNK2","ARVCF","TRMT2A","RANBP1","ZDHHC8","KLHL22","MED15","SERPIND1","SNAP29","CRKL","LZTR1","CECR2","SMARCB1","P2RX6","DERL3","SLC7A4","BCL2L13","DDTL","DDT","OSM","CABIN1","TBC1D10A","SUSD2","SF3A1","GGT5","RNF215","SEC14L2","SEC14L3","SPECC1L","PPIL2","UPB1","YPEL1","SNRPD3","PES1","MAPK1","GGT1","PRODH","PPM1F","SLC35E4","TOP3B","CRYBB3","CYTH4","ESS2","MFNG","CARD10","LRP5L","SLC25A1","GRK3","LGALS2","GGA1","HIRA","SH3BP1","LGALS1","HPS4","PIK3IP1","SRRD","PATZ1","TRIOBP","TFIP11","GCAT","GGTLC2","ANKRD54","EIF3L","SNU13","MICALL1","POLR2F","SOX10","CCDC134","DEPDC5","PICK1","TTC28","SLC16A8","CENPM","SEPTIN3","SLC5A1","KDELR3","CYP2D6","DDX17","DMC1","TCF20","HSCB","CBY1","TOMM22","RSPH14","XBP1","RTCB","JOSD1","FBXO7","GTPBP1","POLDIP3","RAB36","TIMP3","PPP6R2","SBF1","SUN2","CYB5R3","DNAL4","MIOX","LMF2","RHBDD3","PACSIN2","TTLL1","RASL10A","AP1B1","HMGXB4","TOM1","NEFH","CHKB","BIK","HMOX1","MCAT","THOC5","MCM5","ARSA","TSPO","TTLL12","CBX7","PDGFB","RPL3","ZMAT5","RBFOX2","SYNGR1","TAB1","ASCC2","MTMR3","MIEF1","APOL4","PNPLA5","APOL1","PNPLA3","MYH9","SAMM50","TXN2","FOXRED2","GRAP2","EIF3D","TNRC6B","SGSM3","IFT27","KIAA0930","NCF4","CSF2RB","SLC25A17","FAM118A","KCTD17","ST13","IL2RB","RBX1","EP300","L3MBTL2","CHADL","RANGAP1","ZC3H7B","PHF5A","ACO2","POLR3H","TRMU","PMM1","DESI1","CERK","BRD1","ZBED4","MLC1","HDAC10","KCNK10","ABHD4","KHNYN","FKBP3","SDR39U1","GZMH","GZMB","RBM23","PRMT5","COCH","AP4S1","POLE2","VCPKMT","SOS2","CDKL1","NIN","PYGL","TRIM9","PSMC6","GNPNAT1","DDHD1","CNIH1","CGRRF1","ATP6V1D","CCDC198","PLEK2","PIGH","PSMA3","VTI1B","TIMM9","GSTZ1","KIAA0586","TMED8","SAMD15","AHSA1","DAAM1","SPTLC2","RIN3","LGMN","ALKBH1","SNW1","CHGA","ITPK1","DHRS7","PPM1A","SIX4","GALNT16","ASB2","CEP128","ERH","HIF1A","SUSD6","SRSF5","SLC10A1","EIF5","DICER1","ZFYVE21","MTHFD1","TCL1A","ZC3H14","TELO2","PCNX1","BDKRB1","GSKIP","VRK1","PSMC1","PAPLN","RPS6KA5","PPP4R3A","C14orf93","PSMB5","YY1","ACIN1","CCNB1IP1","TRIP11","APEX1","PABPN1","EFS","ARHGAP5","CINP","DHRS2","SRP54","CHD8","PCK2","PRORP","DCAF11","PSMA6","NFKBIA","EMC9","PSME2","BRMS1L","REC8","TM9SF1","SEC23A","GMPR2","PNN","RABGGTA","NFATC4","PLTP","PCIF1","GSS","MMP9","TRPC4AP","PYGB","ABHD12","PROCR","GINS1","NINL","CD40","UQCC1","ZMYND8","SGK2","IFT52","MYBL2","HNF4A","NDRG3","SLA2","RAB5IF","NFATC2","PABPC1L","STK4","SALL4","ADNP","PFDN4","CSTF1","BMP7","RAE1","TPD52L2","DNAJC5","NELFCD","CTSZ","PRPF6","TUBB1","PRELID3B","MTG2","PSMA7","SLCO4A1","MRGBP","TCFL5","DIDO1","GID8","SLC17A9","ARFGAP1","COL20A1","EEF1A2","PTK6","GMEB2","C20orf27","SPEF1","CDC25B","RNF24","ARFRP1","NDUFAF5","TRIB3","RASSF2","CSNK2A1","SLC52A3","ANGPT4","CDS2","HM13","SNPH","SIRPB1","SEC23B","FERMT1","PLCB4","MYL9","HCK","TM9SF4","TLDC2","CRNKL1","POFUT1","SAMHD1","KIF3B","MROH8","NOP56","MANBAL","IDH3B","MAPRE1","JAG1","CDK5RAP1","SNTA1","TTI1","E2F1","RPRD1B","PXMP4","CHMP4B","BPI","CST3","ACTR5","WFDC2","AHCY","PPP1R16B","FAM83D","EPPIN","DHX35","DNTTIP1","MAP1LC3A","PIGU","ACOT8","APMAP","CELF4","ZNF516","ADNP2","RBFA","USP14","VAPA","METTL4","LPIN2","SMCHD1","MYOM1","MYL12A","CEP76","ST8SIA5","CEP192","RNMT","SMAD7","LIPG","LAMA1","RNF125","ANKRD12","POLI","MIB1","RBBP8","RIOK3","CSTF2","MXRA5","VSIG1","PSMD10","ATG4A","STS","TBL1X","GPR143","PGRMC1","POLA1","MID1","NKAP","NXT2","GUCY2F","ATP1B4","ALG13","PRPS2","TLR8","MOSPD1","AMMECR1","WDR13","SUV39H1","XIAP","STAG2","ATP11C","ABCD1","CCDC22","CACNA1F","SYP","PLP2","BMX","PLS3","NAA10","RENBP","ELF4","SMARCA1","MTMR8","ASB9","ZC3H12B","RBBP7","KCND1","SLC25A14","FMR1","PIM2","SLC35A2","PQBP1","RS1","PCSK1N","EMD","TAZ","PGK1","MAGT1","SMS","PHEX","UBL4A","CD99L2","EEA1","RP2","JADE3","CDK16","USP11","PCYT1B","HTATSF1","VGLL1","CD40LG","TIMP1","GABRE","PCDH11X","FGD1","PIN4","PORCN","ITIH6","MAGED2","RBM3","KLF8","SRPX2","SYTL4","ZDHHC15","CENPI","PBDC1","GLA","ARMCX3","BEX4","RUBCNL","NALCN","FGF14","NDFIP2","TNFSF13B","FNDC3A","CDADC1","CAB39L","KLF5","STK24","ACP5","DNAJC3","UGGT2","ARHGEF7","FGF9","PARP4","SUPT20H","MRPS31","SLC25A15","KPNA3","FLT1","RGCC","VWA8","DGKH","KATNAL1","INTS6","ACOD1","DHRS12","TSC22D1","CLN5","MSLN","MGRN1","ZNF629","TRADD","HSF4","CORO1A","MAPK3","GDPD3","ELMO3","PHKB","LYRM1","NUTF2","NUP93","CENPT","TSNAXIP1","NFAT5","LONP2","N4BP1","ARL2BP","PLLP","CCL22","DHODH","CCL17","CTCF","ACD","POLR2C","PARD6A","ZNF821","MMP15","USB1","CYB5B","CCDC113","NME3","NDRG4","PSMD7","SETD6","SLC38A7","VAC14","HAS3","TANGO6","COG4","SMPD3","SLC7A6OS","SLC7A6","PLA2G15","ESRP2","FA2H","WDR59","MON1B","CMC2","AXIN1","HCFC1R1","NPRL3","MLYCD","MPG","NECAB2","HSDL1","TAF1C","NAGPA","WFDC1","SEC14L5","COTL1","USP10","CRISPLD2","TSC2","ZNF500","NME4","ABCC1","NOMO3","LMF1","CIAO3","MTHFSD","CLCN7","HAGHL","ANTKMT","SLC7A5","METRN","FBXO31","STUB1","RHBDL1","NUBP1","UBE2I","ZP2","MEFV","CRYM","EEF2K","CAPN15","PIEZO1","GSPT1","ZNF174","CLUAP1","UBFD1","EARS2","ELOB","GGA2","CPPED1","USP31","HMOX2","DNAJA3","CORO7-PAM16","BFAR","TOX3","RBL2","QPRT","XYLT1","PYCARD","RPGRIP1L","MAZ","STX4","CDIPT","BCKDK","KAT8","NOMO1","IL21R","SYT17","TMC5","CCP110","VPS35L","RNF40","KNOP1","AQP9","AAGAB","IQCH","LACTB","CORO2B","CSK","HERC1","TRIP4","MTFMT","AP3B2","ACSBG1","RAB11A","CTSH","TTC23","CD276","FAH","CEMIP","RPAP1","HOMER2","EHD4","TMEM87A","ZNF106","CEP152","ATP8B4","DTWD1","GABPB1","TJP1","BMF","DMXL2","SCG3","DNAJC17","EIF3J","SPG11","RHOV","VPS18","OIP5","SLC30A4","BLOC1S6","MYEF2","SGK3","PDGFRL","CSPP1","ZDHHC2","BRF2","TRIM35","ZFAND1","RP1","CA2","FZD3","INTS9","RIPK2","EYA1","NBN","TRPA1","CPQ","DECR1","CALB1","IMPAD1","SFRP1","LAPTM4B","UBE2W","POP1","NIPAL2","IKBKB","PLAT","JPH1","STK3","GDAP1","RAB2A","EIF3E","EMC2","ESRP1","CCN4","NDRG1","ZC2HC1A","IL7","ARMC1","TRPS1","SPAG1","CHRAC1","NCALD","SNX16","UBR5","GSDMD","TSTA3","PYCR3","EEF1D","ANXA13","SQLE","SH2D4A","INTS10","ERI1","SLC39A14","MTMR9","LEPROTL1","DCTN6","R3HCC1","GSR","TNFRSF10A","UBXN8","PPP2CB","ERICH1","TUSC3","ARHGEF10","KLHDC4","MCM4","KCTD9","ASAH1","BNIP3L","MAN2B1","KCNN4","TULP2","NUCB1","GYS1","MAP4K1","ECH1","HNRNPL","NFKBIB","TUBB4A","SARS2","KCNA7","SNRNP70","CLPTM1","RELB","CLASRP","LIN7B","PPP1R37","FCGRT","PIH1D1","ARHGEF18","PPP1R13L","PEX11G","ERCC2","DOT1L","PLEKHJ1","SLC17A7","RNASEH2A","KLC3","CD37","SF3A2","AMH","LYL1","OAZ1","TRMT1","STX10","FCER2","DMPK","TBC1D17","IL4I1","CCDC130","PTOV1","TLE5","NOVA2","SGTA","LILRB1","MED25","LILRA1","SNAPC2","C19orf53","TIMM44","CCDC61","IL27RA","ASF1B","TNNT1","VRK3","FAM32A","PPP6R1","C19orf44","MED26","OLFM2","RASAL3","AKAP8","EPHX3","ILVBL","ZNF419","CASP14","SLC1A6","AURKC","POP4","CCNE1","URI1","PDCD5","ANKRD27","RPS16","TIMM50","FBL","DYRK1B","GPI","AKT2","PLD3","PRX","PIAS4","NUMBL","EBI3","YJU2","TBCB","FSD1","POLR2I","CLIP3","ZFR2","SLC1A5","PRKD2","TJP3","APLP1","CACTIN","CCDC9","HNRNPUL1","FZR1","BBC3","TGFB1","DENND3","DMAC2","CEACAM4","PLIN3","MYH14","MRPL4","SIGLEC8","CD79A","ICAM4","RPS19","NOP53","NKG7","ICAM5","ETFB","CD33","CEACAM5","CRX","BABAM1","TYK2","SULT2A1","CDC37","NAPA","RABAC1","ATP1A3","MEIS3","PTPRS","CNFN","MEGF8","KDELR1","CYTH2","GRWD1","GRIN2D","CCDC114","CARD8","LIG1","SIGLEC6","ZNF175","PLA2G4C","SIGLEC5","HAS1","RAB3D","DBP","TMEM205","CAPS","PLPPR2","FAM83E","RASIP1","BCAT2","MIER2","PLEKHA4","PPP2R1A","TNPO2","WDR83OS","GCDH","LILRB5","DNASE2","MAST1","LENG1","PRPF31","TFPT","JAK3","RPL18A","SLC5A5","ARRDC2","PIK3R2","RAB3A","PDE4C","ISYNA1","ELL","CRTC1","COMP","COPE","DDX49","ARMC6","TMEM147","GAPDHS","HAMP","USF2","LSR","KXD1","FKBP8","SUGP1","HPN","ZNF14","SCN1B","PBX4","ERF","GSK3A","ATP13A1","ZNF574","SIPA1L3","ZNF85","ETHE1","CADM4","SMG9","AVL9","RUNDC3B","CFAP69","GTPBP10","RASA4","CDK6","PMPCB","DNAJC2","TFPI2","BET1","NAMPT","TWISTNB","PIK3CG","PON3","PON2","ITGB8","HBP1","DUS4L","SP4","WDR91","DNAH11","CBLL1","MTPN","PTN","MPP6","GSDME","ATP6V0A4","ZC3HAV1","TTC26","OGDH","ADAP1","TFEC","H2AZ2","CAV2","MET","RNF32","LMBR1","DNAJB6","LFNG","BRAT1","IQCE","VIPR2","TSPAN12","SSBP1","CPED1","HIBADH","TAX1BP1","CPVL","CHN2","GRB10","ABHD11","COBL","FKBP14","PLEKHA8","STX1A","NOD1","GARS1","CRHR2","EPHB6","MINDY4","CASP2","CHCHD2","HSPB1","PDAP1","BUD31","PTCD1","CYP3A5","ZKSCAN1","EIF3B","SNX8","NUDT1","PTPRZ1","TAF6","WASL","HYAL4","AIMP2","TFR2","MOSPD3","PCOLCE","FBXO24","RBM28","USP42","IMPDH1","AGFG2","LSM5","SERPINE1","AP1S1","MOGAT3","C1GALT1","PLOD3","RPA3","ZNHIT1","CLDN15","GLCCI1","PHF14","NRF1","TMEM106B","EZH2","CEP41","ZNF862","SFRP4","MEST","ANKMY2","ACTR3C","TSPAN13","RARRES2","AGR2","AHR","CHCHD3","GIMAP2","TMEM176B","GLI3","PSMA2","MRPL32","COA1","BLVRA","URGCP","TMEM248","RHEB","PRKAG2","AEBP1","POLD2","GCK","BCL7B","YKT6","TBL2","CLIP2","EIF4H","LIMK1","SPATA6L","SLC1A1","FKTN","FSD1L","CNTNAP3","SPIN1","NMRK1","TMEM245","PRUNE2","MEGF9","TRIM14","CORO2A","TGFBR1","SEC61B","C5","OGN","ASPN","ECM2","TLE4","LHX6","PTGR1","SUSD1","AKNA","TNFSF8","DNM1","ENG","AK1","CDC37L1","PLGRKT","TBC1D13","RIC1","KDM4C","DOCK8","KANK1","ELAVL2","NCS1","TESK1","KCNT1","FUBP3","CREB3","RGP1","MPDZ","DDX58","EDF1","PIP5K1B","GLIS3","BAG1","RAPGEF1","NPDC1","APBA1","SETX","PTGDS","ABCA2","SHB","UBE2R2","ABHD17B","EXOSC3","ZFAND5","DVL1","PDLIM1","CCNJ","GATA3","HPS1","PHYH","RASSF4","DNMBP","RAB11FIP2","CXCL12","ERLIN1","EIF3A","CUBN","TRDMT1","DDX50","MAPK8","SEC23IP","ATE1","NSMCE4A","PLEKHA1","PALD1","UNC5B","CDH23","VSIR","SPOCK2","MICU1","PPP3CB","CCSER2","BMPR1A","MINPP1","ACTA2","LIPA","TWNK","LZTS2","SFXN3","KAZALD1","FBXW4","FGF8","NPM3","TNKS2","GBF1","ARHGAP21","CPEB3","FBXL15","CUEDC2","SUFU","ANKRD26","ACBD5","LHPP","LARP4B","GTPBP4","EDRF1","BCCIP","MTPAP","NEURL1","SH3PXD2A","PITRM1","STN1","MAP3K8","DKK1","EBF3","GLRX3","SORCS1","TASOR2","XPNPEP1","SMC3","SHOC2","TFAM","CCDC6","CUL2","CCNY","UBE2S","RPL28","ZMIZ1","PPIF","PBLD","TSPAN14","LGI1","TBC1D12","CYP2C18","KRT23","CRYBA1","NUFIP2","GIT1","RPL19","FBXL20","RUNDC3A","UBTF","CSF3","PSMD3","CASC3","RAPGEFL1","RGS9","RNF43","WNT3","ASPA","RAD51C","SEPTIN4","MTMR4","TRIM37","P2RX1","DHX40","TUBD1","KPNB1","GOSR2","PNPO","RPS6KB1","TRIM16L","CDK5RAP3","CBX1","RECQL5","PIGL","GALK1","INTS2","CAMTA2","MED13","ENO3","PFN1","RNF167","SLC25A11","RASD1","CHRNE","RAI1","NUP88","C1QBP","SLC6A4","BLMH","CPD","GOSR1","CCDC47","MED31","DRG2","FTSJ3","AKAP10","ALDH3A1","SMARCD2","ICAM2","SYNGR2","B9D1","UTP6","DDX5","C17orf75","CYTH1","PSMD11","LGALS3BP","CCL2","CCL8","PEX12","DHX58","KAT2A","RAB5C","NAGLU","HSD17B1","MLX","CNTNAP1","ABI3","EZH1","DLX4","PPP1R9B","COL1A1","MRPL27","VAT1","LRRC59","RND2","ALOX12","HDAC5","ABCC3","LUC7L3","SMURF2","DUSP3","EFTUD2","HLF","SLC16A6","PRKAR1A","EFNB3","FAM20A","YWHAE","MMD","RANGRF","DPH1","MAP2K6","DHRS7B","WSB1","RCVRN","SLC9A3R1","MYH3","NAT9","TMEM104","TNFAIP1","IFT20","TMEM97","CDR2L","PMP22","UNC119","ALDOC","SUPT6H","RAB34","PHF12","TMEM33","GABRA4","SLAIN2","OCIAD1","CWH43","DCUN1D4","USP46","SULT1E1","CHIC2","NMU","CRACD","LAMTOR3","NFKB1","AREG","MANBA","UBE2D3","ELF2","NDUFC1","TBC1D9","ZNF330","INPP4B","GAB1","KLHL2","CPE","RPL34","WFS1","ANXA10","GRPEL1","GAR1","FRG1","CLCN3","AADAT","GALNT7","DHX15","SEPSECS","TRIM2","SLC2A9","FBXW7","NEIL3","TBC1D19","CLNK","NSD2","SH3D19","STIM2","MFSD10","GLRB","BST1","RAPGEF2","HGFAC","SNX25","LRP2BP","UFSP2","KLF3","KLHL5","FAM149A","NCAPG","UGDH","PPARGC1A","DDX25","HTATIP2","CTSC","CCDC34","ZBTB16","ELP4","ZPR1","MTCH2","FNBP4","TECTA","SC5D","CRTAM","JHY","B3GAT1","HSPA8","VWA5A","DNAJC4","SIAE","SNX15","LPXN","DTX4","ATG2A","EHD1","OSBP","UNC93B1","PUS3","DCPS","KMT5B","FOXRED1","PPP6R3","MS4A6A","MS4A4A","ST3GAL4","CPT1A","CCND1","CCDC86","PRPF19","TMEM109","HPX","TRIM3","CHORDC1","FOLR1","ANAPC15","PANX1","ARHGEF17","CEP164","RNF141","CEP126","EIF4G2","IL10RA","GALNT18","BIRC2","UBE4A","DDX6","CBL","NECTIN1","HIPK3","KIAA1549L","FBXO3","PDHX","SLC1A2","COMMD9","SLC15A3","CD5","ACCS","MDK","AMBRA1","MADD","PTPMT1","NAA40","CARS1","SLC22A18","CD81","SLC35F2","C11orf21","ELMOD1","SOX6","C11orf58","PITPNM1","RPS13","AIP","NUP98","NDUFS8","TCIRG1","CHKA","EXPH5","HPS5","GTF2H1","POU2AF1","PTPN5","VWF","PSMD9","P3H3","PPFIBP1","PRPF40B","CD69","PRDM4","CLEC2B","COQ5","SELPLG","CORO1C","ASIC1","CAPRIN2","TSPAN11","KCTD10","SLC11A2","MLEC","MVK","CSRNP2","CAMKK2","BIN2","IL23A","ATP5F1B","PTGES3","BCL7A","RSRC2","CYP27B1","LIN7A","KRT18","ACSS3","TNS2","GLI1","PPM1H","METAP2","LTA4H","ELK3","SLC6A12","WNT5B","MAGOHB","TRPV4","ITFG2","FOXM1","PRH1","PRMT8","PARP11","ARPC3","GPN3","VPS29","RAD51AP1","SH2B3","AKAP3","MANSC1","KCNA1","DUSP16","CREBL2","ACAD10","ALDH2","CDKN1B","GPRC5D","NAA25","GSG1","SCNN1A","LTBR","OGFOD2","CDK2AP1","OAS3","OAS2","ART4","MGP","RASAL1","ARHGDIB","GTF2H3","EIF2B1","DDX55","SLC38A1","ENDOU","C12orf49","RFC5","STX2","ADGRD1","COPZ1","TBC1D30","CAND1","IFNG","RAB5B","MDM1","NUP107","CNOT2","TIMELESS","CPSF6","KRR1","MRPL51","GAPDH","NOP2","CHD4","ACRBP","UHRF1BP1L","COPS7A","ING4","GNB3","CDCA3","CHPT1","USP5","TPI1","GNPTAB","SPSB2","ENO2","ATN1","C12orf57","PTPN6","LPCAT3","NT5DC3","SLCO1B3","NANOG","SUDS3","GOLT1B","PRKAB1","CMAS","HCFC2","ST8SIA1","CLEC4A","C2CD5","AICDA","RAB35","PHC1","COX6A1","RFX4","RIC8B","SRSF9","FGFR1OP2","KLRB1","COL12A1","BTN3A3","TDP2","FRK","DSE","RWDD1","RSPH4A","MAK","TMEM14C","PAK1IP1","GCNT2","SMIM8","NEDD9","CEP85L","ADTRP","ASF1A","MCM9","FAM184A","RNGTT","MAN1A1","GABRR2","SERINC1","HDDC2","TPD52L1","HINT3","NCOA7","RIPOR2","SASH1","UST","ULBP1","FBXO5","MTRF1L","PPARD","OPRM1","FANCE","TULP1","SLC26A8","MAPK14","KCTD20","STK38","SRSF3","SOD2","MRPL18","MCM3","RNF8","PHACTR1","MDGA1","ICK","FBXO9","CD83","MDN1","SAYSD1","BACH2","RBM24","CAP2","TREML2","ZNF451","BAG2","RAB23","GPR63","KHDRBS2","FBXL4","CCNC","E2F3","PTP4A1","ASCC3","BVES","MED23","WASF1","GPLD1","ALDH5A1","CRYBG1","VNN1","VNN2","ACOT13","SMAP1","RPS12","C6orf62","B3GAT2","GMNN","EYA4","SOBP","NR2E1","SNX3","HBS1L","TRIM38","PEX7","ZBTB24","FIG4","PERP","ARFGEF3","SLC16A10","HECA","ADGRG6","PHACTR2","EPM2A","SLC39A7","CCR6","SLC22A2","PHF1","CUTA","PACRG","QKI","C6orf118","PDE10A","MDFI","TFEB","CCND3","BYSL","FAM120B","TBP","GUCA1B","PRPH2","BICRAL","PPP2R5D","MRPL2","PTK7","SRF","CUL9","DNPH1","DUSP22","EXOC2","COX7A2","TMEM30A","GMDS","SENP6","IMPG1","PRPF4B","TTK","SLC29A1","CCN6","BTN2A1","LAMA4","TENT5A","CLIC5","FBRSL1","ENPP5","LY86","PRSS16","TBX18","ERBIN","PCDHB2","HARS2","NUDT12","CEP72","MAN2A1","SEMA5A","TENT4A","GHR","HMGCS1","DAP","NME5","BRD8","KIF20A","NNT","MRPS30","HSPA9","MRPS27","PFDN1","HBEGF","SLC4A9","LOX","CDH9","APBB3","TMCO6","SPARC","IK","HMGCR","CERT1","FAF2","PDE8B","CLK4","PCDHB15","HAVCR1","GRM6","ITK","RNF130","THG1L","ARSB","CLINT1","THBS4","CNOT6","IL12B","BTNL8","TTC1","MSH3","RASGRF2","GABRG2","CCNG1","POLR3G","DROSHA","CDH6","LMNB1","ARRDC3","GOLPH3","SUB1","NPR3","FAM172A","TARS1","LNPEP","PDE4D","RAD1","BRIX1","PRLR","SLC12A7","RAD50","ST8SIA4","GNPDA1","PCDH12","SKP1","NUP155","PPP2CA","FGF1","NR3C1","C5orf15","PPWD1","LIFR","TRIM23","TRAPPC13","SEC24A","TXNDC15","TTC33","RARS1","WWC1","MACROH2A1","TCERG1","DPYSL3","SMAD5","CSNK1A1","HMGXB3","ERGIC1","PDGFRB","ATP6V0E1","BNIP1","CPEB4","HRH2","DBN1","ZNF346","UNC5A","EHHADH","CNTN3","SMC4","SELENOK","ACTR8","TBCCD1","TIMMDC1","CRBN","HRG","BCL6","HGD","CLDN16","ARL6","NPHP3","CD86","AMOTL2","NIT2","FAM162A","OGG1","KPNA1","PCCB","UBE3A","ARMC8","CEP70","RBP2","RBP1","SLC25A36","GRK7","RNF7","TFDP2","XRN1","KAT2B","SERPINI2","PDCD10","WNT5A","PFKFB4","COL7A1","FGF12","PRKAR2A","HES1","USP4","ACAP2","ECT2","GNAT1","GNAI2","TFG","USP9Y","HYAL1","TUSC2","NPRL2","RPL24","CYB561D2","C3orf14","FXR1","CBLB","BBX","IFT57","GNB4","HHLA2","IQCG","GBE1","UMPS","NCBP2","SNX4","C3orf52","FRMD4B","SLC41A3","ROPN1B","PLXNA1","ATP6V1A","ABTB1","PODXL2","UPK1B","CSPG5","KLHL18","SCAP","EFCC1","KIAA1257","NEK11","MRPL3","PLSCR4","HEMK1","CISH","MAPKAPK3","ACVR2B","WDR48","COMMD2","GORASP1","PEX5L","RRP9","ABCC5","AADAC","ABHD14B","EIF1B","ABHD14A-ACY1","ARHGEF26","KLHL24","PLCH1","VIPR1","DNAH1","SSR3","ZBTB47","NKTR","CLCN2","FOXP1","EIF4G1","SPCS1","NEK4","SLC4A3","INO80D","EEF1B2","DGUOK","MOB1A","KANSL3","LMAN2L","RTKN","TTL","IL1A","CCL20","PIKFYVE","KCNIP3","FAHD2A","NCL","ACTR1B","SLC35F5","ZAP70","ACTR3","STEAP3","EPB41L5","TFCP2L1","SF3B6","TP53I3","DNAJC27","STAM2","OTOF","GPD2","CYTIP","ACVR1","TANC1","SLC30A3","MPV17","GTF3C2","EIF2B4","NRBP1","ITGB6","ITGA4","PSMD14","SNX17","ASB3","PPM1G","PDE1A","REEP6","PCSK4","APC2","IFIH1","RPS15","GCA","INO80B","MOGS","TTC31","NDUFS7","PCGF1","CLIP4","SPTBN1","AUP1","RTN4","HTRA2","LOXL3","DOK1","GALNT3","POLE4","TACR1","CCDC88A","ACADL","EVA1A","MRPL19","LANCL1","WDR75","EFEMP1","FANCL","FN1","STAT1","GLS","PAPOLG","DNAH6","PECR","UNC50","IGFBP2","ELMOD3","IGFBP5","USP34","EFHD1","KCNJ13","CCT4","GGCX","EHBP1","OTX1","TXNDC9","COQ10B","GNLY","SF3B1","ST3GAL5","CHST10","PDCL3","MOB4","HSPE1","KDM3A","PLCD4","CHMP3","ZNF142","IL1R2","IL1R1","IL1RL2","IL1RL1","IL18R1","IL18RAP","SLC9A2","FHL2","MLPH","CNPPD1","UXS1","ABCB6","STK16","HDLBP","PPP1R7","PASK","STK25","TPO","PROC","ID2","TAF1B","HPCAL1","ODC1","BIRC6","NOL10","PLEKHB2","GORASP2","STRN","CEBPZ","PRKD3","DCAF17","QPCT","RAB3GAP1","SLC25A12","RMDN2","LCT","DARS1","SRSF7","SDC1","PLCL1","SLC1A4","SOS1","KYNU","WIPF1","ORC2","COX7A2L","PNO1","ORC4","PLEK","RND3","ATF2","THADA","AAK1","TRAK2","C2orf42","TIA1","PCYOX1","EPAS1","ARID3A","SUMO1","CD207","GRIN3B","ATP6V1B1","NFE2L2","MSH6","PLEKHA3","SPR","EPHA4","PARD3B","FARSB","ALMS1","BCL9","PRRX1","DHCR24","DNAJC16","MARK1","TNR","MORN1","GPX7","CACYBP","SCP2","PAPPA2","RALGPS2","ANGPTL1","CEP104","FAM20B","TCEANC2","TMEM59","LRRC42","WRAP73","MRPL37","ICMT","RPL22","CHD5","QSOX1","STXBP3","PHF13","ERRFI1","PARK7","KIAA1324","OPRD1","AMPD2","SRSF4","MECR","KCNC4","EDEM3","WDR77","ATP5PB","RAP1A","HDAC1","CAPZA1","S100PBP","RNF19B","SCAMP3","TRIM62","ASH1L","DLGAP3","SFPQ","RHOU","GON4L","ARHGEF2","LAMTOR2","MEF2D","DOCK7","SRM","FBXO2","FBXO6","C1orf21","SWT1","MAD2L2","DNAJC6","LEPR","IVNS1ABP","KIAA2013","MFN2","PRG4","MIIP","SMG7","NCF2","PDC","SLC35D1","PLA2G4A","GADD45A","WLS","PRDM2","RGS2","RO60","AMPD1","UCHL5","BCAS2","SRSF11","CTH","AGMAT","OLFML3","TNNI3K","CFHR3","PLEKHM2","CRYZ","PHTF1","ZBTB17","CD58","TFAP2E","CD2","TTF2","NR5A2","KIF21B","TMEM9","ADPRHL2","MAP7D1","WARS2","OSCP1","MRPS15","EXOC8","GNPAT","TSNAX","C1orf109","RRAGC","LGALS8","MTR","BMP8B","MYCL","SIPA1L2","RLF","KMO","ZNF684","RIMS3","AKT3","ETV3","ACADM","SLAMF1","CD48","ADGRL2","PADI2","SDHB","MFAP2","RPF1","KDM5B","UAP1","CTBS","RGS4","KLHL12","SSX2IP","ZNHIT6","PLA2G2D","RBBP5","GBP3","GBP1","KIF17","GPR89A","CDK18","RAB29","CD160","ECE1","HMGCL","GALE","ID3","CR2","CD46","PRPF3","APH1A","P3H1","SLC2A1","EBNA1BP2","CDC20","MPL","IPO13","ATP6V0B","B4GALT2","ERI3","PTCH2","AKR1A1","PRDX1","PIK3R3","TSPAN1","BLZF1","CCDC181","SLC19A2","FAAH","NSUN4","TMED5","MROH9","DR1","CNN3","PRRC2C","F3","ABCD3","VAMP4","DPH5","FASLG","PTBP2","TNFSF4","PRDX6","DARS2","HSD11B1","IRF6","UTP25","SERPINC1","RCAN3","SYF2","RSRP1","SLC35A3","RCOR3","STMN1","MTFR1L","MAN1C1","NEK2","RPS6KA1","DHDDS","NENF","NSL1","PROX1","ARID1A","CENPF","RPA2","PPP1R8","STX12","MARC2","SLC5A9","OSBPL9","TXNDC12","ESYT2","CD3EAP","MESD","RCN2","CHRNB4","MUC5B","CTSD","COLEC11","STAG1","STK11","KMT2A","IFT46","SLC8A2","KPTN","RPS25","KIF14","TNNT2","DDX59","CAMSAP2","ATF6","MREG","FASTKD2","NRP2","CREB1","KLF7","TTR","B4GALT6","C1orf54","CA14","CASC1","LRMP","ATP10B","SPCS2","USP35","FILIP1","CASP8AP2","HMGN3","UBE3D","ANKRD13C","SGIP1","PHF3","ADGB","PLAGL1","FBXO30","TNFAIP3","AKAP7","RAB32","MYB","ALDH8A1","SGK1","RNF146","ARG1","CCN2","PMFBP1","FBXL5","MED28","SLC16A7","RXYLT1","ZNF430","VAMP8","DCLRE1B","MYL12B","FOXO3","ARMC2","RPN2","TGIF2","CASQ2","PKD2","ABCG2","SPP1","STBD1","CCNI","RARRES1","MFSD1","RAB3GAP2","EEF2KMT","PPL","UBN1","KLF12","UCHL3","PCDH17","HS1BP3","LDAH","WDR35","CCND2","FGF23","ELL2","DNAH7","CYP20A1","NDUFB3","GTF3C3","SATB2","UBE2B","TRPM6","GDA","KLF9","TJP2","ECRG4","ITGB1BP1","CPSF3","PIGZ","SENP5","CCDC92","C1orf198","HEATR1","PTBP3","RAD23B","FKBP15","CTNNAL1","ABITRAM","WDR34","SET","PTPA","GLE1","RAB14","CNTRL","TRIM32","FBXW2","PHF19","NEK6","BSPRY","PPP6C","NDUFA8","HDHD3","RBM18","HSDL2","MAPKAP1","NR4A3","INVS","GALNT12","DENND1A","ALG2","CSF3R","KDSR","VPS4B","C19orf25","ZBTB45","YLPM1","DCAF4","PROX2","FCF1","PGF","IFI27L2","BBOF1","NEK9","ACYP1","IFT43","NPC2","DNAL1","IRF2BPL","ACOT2","LTBP2","AREL1","MLH3","TTLL5","FLVCR2","ABCD4","DLST","PPP4R4","TGFB3","ZC2HC1C","SLIRP","RBM25","ALDH6A1","GPR68","EIF2B2","NRDE2","COQ6","ZNF410","RHOQ","GPR75","SUPT7L","KLHL29","DNMT3A","TMEM214","ATAD2B","FKBP1B","ATL2","YPEL5","FAM98A","YIPF4","AFTPH","LGALSL","CNRIP1","BCL11A","CRIPT","EPCAM","SLC17A5","OGFRL1","SLF2","IDE","IFIT3","IFIT2","GPAM","CUTC","PPP1R3C","PYROXD2","CNNM1","MXI1","SMNDC1","C10orf88","HELLS","PRLHR","TCTN3","DENND10","AVPI1","WDR11","ARMH3","KCNIP2","CFAP58","GOT1","C10orf95","GNA13","KANSL1","HOXB3","DUSP1","PANK3","RCL1","CAAP1","MOB3B","INSL6","INSL4","MLANA","CD274","NUP43","MTHFD1L","LRP11","CCDC170","PCMT1","PLEKHG1","MYCT1","CXorf21","CYSTM1","WDR55","ARAP3","PCDHB14","MRPS14","CENPL","SEC16B","GORAB","ACAT2","TCP1","SNX19","KCNJ5","MSANTD2","TP53AIP1","ARR3","PDZD11","SLC10A7","NUDCD1","ENY2","MASTL","KIAA1217","PLXDC2","EPC1","IQSEC3","CCDC77","TAF12","ENOX1","MTRF1","SOHLH2","DNAJC15","PROSER1","UFM1","WBP4","ELF1","SMAD9","HSPH1","KBTBD7","ALG5","EXOSC8","ETF1","TGFBI","FAM53C","SIL1","PAIP2","MYOT","KDM3B","EGR1","SERP1","PLS1","ZFP30","NR2C1","UTP20","TMPO","ARL1","GLT8D2","MTERF2","SOCS2","NFYB","WASHC3","APAF1","DUSP4","CLU","TNFRSF10B","SORBS3","PTK2B","CHRNA2","PPP3CC","PDLIM2","EPHX2","RNF170","UBIAD1","TARDBP","TNFRSF8","ZNF706","LYPLA1","CRISPLD1","COPS5","RDH10","EPX","AKAP1","COIL","TRIM25","SCPEP1","SPOP","SLC35B1","TBX4","TEX14","FAM117A","NCAPH","LRAT","TMEM131L","MND1","TRIM6","ABCC11","TENT4B","ADCY7","CEP89","TSHZ3","ECHDC2","PLBD1","PRB2","PYROXD1","BCL2L14","PSPC1","ZNF549","A1BG","ZSCAN18","ZNF211","RGSL1","LHX4","RNF2","TRMT1L","SEC22A","CSTA","DPPA4","POPDC2","B4GALT4","NAA50","CD80","KIF18A","DESI2","MAPK8IP1","CRY2","PEX16","CAT","PILRB","ZMYM2","TBC1D15","ADGRB2","ZCCHC17","FABP3","KHDRBS1","TMEM39B","CCRL2","CCR2","POLR3GL","GHSR","TNFSF10","ZNF639","PIK3CA","PDS5A","TMEM156","LIAS","CPXM2","TMEM54","ZSCAN20","CSMD2","HPCA","LRIF1","CLCC1","GPSM2","GTDC1","CXCR4","ZRANB3","ACVR2A","POLK","SV2C","FLT3","RPL21","MTIF3","GTF3A","RASL11A","UBL3","FYTTD1","MTERF4","SASH3","OCRL","MRPS2","FMOD","LAX1","PLG","KIAA1191","COPA","CD244","LY9","HS3ST2","RBBP6","ZC3H7A","SERAC1","ANXA11","LDB3","SHLD2","PRXL2A","ZNF205","NAA60","RPL5","ODF2L","PTGFR","SPATA1","TRMT13","LRRC39","RWDD3","ZNF644","CCDC18","RPAP2","SLC66A2","BBS9","PMS2","ZMIZ2","SEPTIN7","EEPD1","KLHL7","HERPUD2","CBX3","HNRNPA2B1","WIPF3","FAM126A","INHBA","FKBP9","NT5C3A","ARL4A","CCZ1","POLM","MRM2","SMU1","GLIPR2","SLC25A51","CLTA","RECK","ACO1","PHF24","DNAI1","DCAF10","CNTFR","KIAA1549","TRIM24","CYREN","CALD1","AKR1D1","PLAU","SRGN","CHST3","BICC1","CISD1","EGR2","ECD","P4HA1","SLC25A16","ZWINT","VPS26A","RBM19","CIT","IFT81","ACADS","HVCN1","DDX54","MED13L","CDKN2C","RNF11","RASSF8","BHLHE41","SSPN","ITPR2","CCDC91","NECAB1","WWP1","ACOT9","PRDX4","DDX39A","PKN1","TRIR","ADGRE5","WDR83","GIPC1","SPRYD7","EBPL","ATP7B","ZC3H13","NLN","CENPK","OPTN","ITIH5","ATF1","TSFM","ARHGAP9","NCKAP1L","MMP19","PFDN5","SPATS2","ORMDL2","NR4A1","PDE1B","CDK2","LRP1","ATG101","NFE2","IKZF4","SMUG1","TUBA1B","EEF1AKMT3","KBTBD4","SARDH","ATPAF1","STIL","HJURP","COL10A1","NDUFAF4","USP45","MORF4L2","FAM199X","RAB9A","METTL8","TTC21B","NMI","TNFAIP6","BAZ2B","SLC36A1","LPGAT1","BATF3","G0S2","KCNJ2","RAP2C","EXOSC9","PLA2G12A","B9D2","COQ8B","PFKFB2","C4BPA","C4BPB","RAB38","GPR83","AGO2","MXD4","CKS2","DAW1","ACSL3","CHPF","DNPEP","OBSL1","FAM124B","SLC12A4","ENKD1","GCNT7","CTCFL","FAM210B","PI3","FAM209A","SNX21","SLPI","WFDC3","TTPAL","PREX1","KCNS1","SLC12A5","ARHGAP40","SDC4","NCOA3","PIGT","NCOA5","VAPB","PARD6B","ATP5F1E","CHD6","PLCG1","TOX2","SRSF6","GDAP1L1","ARFGEF2","ZNFX1","ZNF831","CSE1L","TMEM189-UBE2V1","RAB22A","PTGIS","STAU1","CDH26","SNAI1","MOCS3","STX16","PMEPA1","RNF114","DDX27","C20orf85","BCAS4","TP53TG5","ZBP1","NEURL2","MAGEA10","MTRR","FASTKD3","PEPD","IQSEC2","VAMP7","IL9R","XG","STAMBP","NAGK","MCEE","PAIP2B","SNRNP27","MPHOSPH10","IL17C","ATP8A1","USP22","POF1B","HIF3A","ZNF576","ZNF45","LYPD3","CEACAM8","USP9X","F13A1","GRM4","TRERF1","PACSIN1","BTN2A2","SIRT5","MRS2","WRNIP1","RRP36","SNRPC","SERPINB6","XPO5","ABCC10","H1-3","PEX6","NQO2",null,"OARD1","UNC5CL","AARS2","H1-1","ZNF391","RPS10","MOCS1","H2BC11","MED20","TBCC","SPDEF","TCP11","MAD2L1BP","APOBEC2","KLHDC3","GNMT","TREM1","MEA1","KLHL31","COL21A1","CDKN1A","SOX4","GLO1","CPNE5","RREB1","SSR1","RIOK1","SLC35B3","RPP40","ATXN1","NUP153","DEK","EEF1E1","RUNX2","LRRFIP1","RAB17","CXCL6","EREG","MYRF","SCGB2A1","AHNAK","EMC3","SSUH2","SH3TC1","CNOT1","LRRC29","BBS2","MT1G","MT2A","C16orf70","GOT2","DOK4","GPR18","CLYBL","TMTC4","RAP2A","ABCC4","EFNB2","SOX21","TM9SF2","HROB","KIF25","IRF1","UPF3B","RNF113A","SEPTIN6","TMEM255A","NDUFA1","DMAC2L","BMP4","PTGER2","FAM193A","GRK4","SOX9","TEKT3","HS3ST3B1","SLC25A35","MRPS7","GGA3","ARMC7","NUP85","SLC25A19","MIF4GD","NT5C","MSTO1","C1orf61","TTF1","GTF3C4","DDX31","PPP1R12C","MBOAT7","SRMS","SLC2A4RG","FNDC11","PPDPF","IL1B","PLGLB2","CHCHD5","PAX8","INSIG2","POLR1B","CCDC93","PSD4","SLC25A23","PSPN","GTF2F1","ALKBH7","CLPP","TNFSF9","THOC2","MED1","RPL23","ATG4C","C3","SH2D3A","TRIP10","GPR108","TNFSF14","FOSB","OPA3","SNRPD2","RTN2","EML2","VASP","SYMPK","GPCPD1","SDCBP2","PANK2","TGM3","CD93","GZF1","NAPB","CENPB","PSMF1","DTD1","RBCK1","TMX4","STK35","SNRPB","NRSN2","AP5S1","RRBP1","ZNF133","FLRT3","OVOL2","PCSK2","MKKS","BFSP1","DSTN","SNRPB2","MGME1","TBC1D20","ITPA","MCM8","TMEM74B","FAM110A","MRPS26","S1PR4","NCLN","HNRNPR","ZNF436","MAX","ARMCX5","MMP24","NECAB3","ID1","RALY","DYNLRB1","EIF2S2","ERGIC3","ROMO1","FAM83C","BPIFB1","CEP250","PLAGL2","MMP24OS","KDM5C","AMOT","TMEM115","PSMB2","AGO3","UROD","ST3GAL3","TMEM53","HECTD3","KLC1","XRCC3","TUBGCP3","MCF2L","PCID2","LRFN3","IGFLR1","CAPNS1","PDCD2L","RBM42","UBA2","FFAR2","HCST","FFAR1","COX6B1","THRA","CCR7","NR1D1","FRMD8","PRDX5","BCL2L12","IRF3","PRMT1","RRAS","PRRG2","SCAF1","PRR12","FLRT1","ASL","SBDS","STATH","HTN1","STAT5A","WNK4","BECN1","PRKCG","TRAP1","GLIS2","NSRP1","DNAJC8","AHDC1","IFI6","ZNF384","EMG1","UXT","CFP","ELK1","TIMM17B","PCNX4","ATG14","KTN1","SIX1","RHOJ","DLGAP5","L3HYPDH","HSPA2","ZBTB1","TRMT5","SGPP1","PLEKHG3","PZP","PRDM7","RHOT1","EVI2A","OMG","WDR60","AIF1L","FAM78A","NUP214","AVPR2","SLC10A3","MAP2K2","HNRNPH2","ARMCX1","TIMM8A","ZC4H2","CANX","INTS11","RGS13","IPPK","ZNF484","FGD3","HIVEP3","PPCS","BCL11B","COX7C","TRAF2","ABHD8","MASP1","ATP13A4","HELB","RAP1B","TSPAN8","BEST3","RAB3IP","PTPRB","DYRK2","YEATS4","TAS2R3","TAS2R4","TAS2R5","LRRC61","IDUA","FGFRL1","TMEM175","AUNIP","PIN1","FBXL12","EMC1","UBR4","HP1BP3","ADGRE2","SIN3B","SLC35E1","EPS15L1","KLF2","OR7C1","F2RL3","UQCR11","GFER","SYNGR3","PKMYT1","WFIKKN1","WDR24","FBXL16","CHTF18","MACF1","SMARCA4","KDM4B","TICAM1","METTL25","EMC6","METTL16","TUBA4A","VIL1","AAMP","PNKD","TNFRSF19","RNF6","ECHS1","ZNF835","AKAP9","SEM1","HIP1","PTPN12","POR","FGL2","STYXL1","STEAP4","GNAI1","PEX1","MTERF1","SGCE","RBM48","CASD1","ZNF780B","LRFN1","ZFP36","SRD5A3","PAICS","PPAT","TUBGCP6","ADM2","DGCR6L","DGCR8","ASPHD2","SDF2L1","YWHAH","GNAZ","MGAT3","ADORA2A","ATF4","A4GALT","CDC42EP1","APOL3","MCHR1","TPST2","BAIAP2L2","MPST","TST","APOL2","RAC2","LIF","C22orf23","APOBEC3A","APOBEC3F","RIBC2","KRT17","EMC4","SPECC1","CPA4","DOCK4","POT1","ATP6V1F","LSM8","CDHR3","PRKRIP1","PODXL","FOXP2","STRIP2","IFT22","MKLN1","DNAJB9","FLNC","LRRC4","CALU","CCDC136","SMO","IRF5","LRRC17","KLHDC10","NDUFA5","FEZF1","MRPS12","MYO1B","MTX2","PDE11A","CHN1","GAD1","OSGEPL1","ORMDL1","HAT1","HERC2","SNRPN","PSMG2","TWSG1","ARHGAP22","WDFY4","EIF2AK4","MYO5C","CGNL1","TMOD2","TTBK2","ELL3","CCDC32","INO80","ICE2","DLL4","ALDH1A2","MINDY2","IVD","KNSTRN","DUT","CHAC1","CLN6","ARPP19","VPS13C","CALML4","ISLR","THAP10","LOXL1","ACKR4","ANAPC13","MBD4","COPB1","PSMA1","SUMF2","PALLD","SPCS3","BBOX1","SERGEF","KCNC1","TPH1","CSRP3","E2F8","DCTD","SOX15","PIMREG","RPAIN","USP6","PLD2","AIPL1","CD68","TXNDC17","ATP1B2","FXR2","KIF1C","MPDU1","PHF20L1","LRRC6","CCNT1","PUS7L","KRI1","ILF3","SLC44A2","AP1M2","CDKN2D","MTUS1","SIGLEC9","KLK10","NGDN","RIPK3","ADCY4","RAB2B","BCL2L2","AJUBA","DTD2","PARP2","HEATR5A","FOXA1","SNX6","EAPP","EGLN3","MIS18BP1","NRL","RNASE1","NEDD8","DAD1","TEP1","EPB41L4A","REEP5","ITFG1","QRICH2","FOXJ1","SEC14L1","RHBDF2","AANAT","ARHGEF6","MAP7D3","FGF13","ASH2L","TTI2","ART1","CHRNA10","CDKN1C","SGO1","RPS4Y1","KLF16","PGAP6","DOHH","MAU2","SHC2","PLPPR3","ABHD17A","SYT5","TNNI3","CBFA2T3","GAMT","PUDP","ERMARD","PHF10","PRRG3","CRACR2A","NXNL2","STARD8","GDPD2","SAT1","GNL3L","SH3BP4","MOSPD2","DOCK6","ECSIT","LDLR","ELOF1","TSPAN16","PRKCSH","CNN1","CDC16","THEM6","NECTIN2","APOE","TOMM40","APOC1","GADD45G","DPP6","XPO7","ACE2","FAM98C","SAFB2","RPL36","ATP8B3","KIF1A","GTPBP3","PLVAP","BST2","SLC27A1","NSUN5","USHBP1","COLGALT1","DDA1","MRPL34","PGLS","LSM7","TULP4","SNX9","RTN4IP1","QRSL1","MTRES1","RSPH3","MAS1","MLLT1","AFDN","ACTN4","STK33","NDUFA10","ARPC1B","CACNG6","ZSWIM6","FCHO1","UNC13A","MAP1S","KLHDC7B","PXDN","SSBP4","GDF15","PGPEP1","IQCN","LSM4","JUND","HRC","TRPM4","ZNF557","CRB3","CAMSAP1","UBAC1","SAG","ZBTB46","HELZ2","SAMD10","LSP1","TNNT3","TNNI2","COL5A1","ATXN10","TUBGCP2","CYP2E1","PNPLA7","PAK4","ZNF337","CEP85","TAF4","RBBP8NL","LAMA5","OSBPL2","ADRM1","ASS1","EXOSC2","POMT1","UCK1","PRRC2B","CHMP2A","UBE2M","TRIM28","METTL26","YIPF2","ATG4D","EIF2S3","TMEM160","ZC3H4","NPAS1","GMFG","MAP3K10","ARHGEF16","LRRC47","SESN2","SMPDL3B","ATP5IF1","MED18","THEMIS2","CLIP1","CCDC62","HIP1R","ZNF317","PPAN","EIF3G","SHFL","DNMT1","ZNF426","SLC6A8","PNCK","DKC1","PLXNA3","MPP1","ZNF331","ZNF236","LRP3","C12orf65","NOL11","UBE4B","CASZ1","PKDREJ","HSD17B3","HABP4","SLC35D2","PRRG1","UBA1","RGN","POLN","PPIL4","AKAP12","SYNE1","ULBP3","LATS1","EPS8L1","LILRB2","AAR2","TTLL9","BPIFA2","RBM39","ZNF341","GGT7","ACSS2","ARHGEF9","GFAP","ATP6V1E1","ZNF227","ZNF428","ZNF141","COX4I1","EMC8","GSE1","GINS2","CHMP1A","SH3BGRL","COX7B","F12","PRR7","NFATC1","IDO1","GJA9","CAP1","PPT1","RAB11FIP4","RLIM","ABCB7","TRAF3","HAUS8","ADGRE3","MRPS25","SH3BP5","HACL1","TBC1D5","CAPN7","RFTN1","C3orf20","RBSN","GALNT15","SLC6A6","KCNC3","NAPSA","NR1H2","PDLIM4","KIF3A","MGAT1","GFPT2","TUBG1","PSME3","RPL27","PSMC3IP","AOC3","ACLY","VPS25","AOC2","G6PC","NDUFA2","ANKHD1","DIAPH1","NDFIP1","UBE2D2","EXOC4","ACAP3","C1orf159","ANO1","PPFIA1","TMEM204","THOC6","TRAF7","NINJ1","CA6","NPHP4","MAP1B","RHOXF2","IL13RA1","WDR44","CKMT2","ZCCHC9","TNS4","TOP2A","STARD3","RARA","PPP1R1B","KHDRBS3","CHD1L","PEX11B","FMO5","PIAS3","PRKAB2","PDHA1","RAI2","MCCC2","ZNF304","ZSCAN5A","ZNF132","USP29","SELENOS","CHSY1","SNRPA1","LLGL1","LIN28A","THAP1","RHPN2","C19orf12","FAAP24","LRRC9","ACTR10","ABHD12B","GCH1","LGALS3","PODNL1","DNAJB1","ZSWIM4","FBXW9","RFX1","ZNF20","C19orf57","DCAF15","CC2D1A","TRIM21","SPATA6","LRRC41","CCT6B","DHX30","RAF1","PPARG","NUP210","FCRLA","HSD17B7","ENOSF1","EMILIN2","SLX1A","ARFIP2","TRIM5","CNGA4","TRIM22","RRP8","TIMM10B","EFR3A","HHLA1","PTCD3","IMMT","MRPL35","IQCA1","ILKAP","PER2","RAMP1","SCLY","PTPRE","RAN","PRKAA1","CARD6","RAP1GAP2","CLUH","INPP5K","MYBBP1A","RPA1","SERPINF1","UBE2G1","EEFSEC","TBC1D14","TMEM128","COQ3","PNISR","SEC61G","LANCL2","FIGNL1","GRSF1","ENAM","JCHAIN","ANKRD17","UTP3","ITGB4","WBP2","H3-3B","UNK","TRIM47","ZRANB2","EIF5A","KDM6B","CLEC10A","SLC52A1","GPS2","XAF1","DLG4","RIDA","VPS13B","RGS22","MATN2","REEP2","PCBD2","SDF2","FLOT2","ERAL1","PRMT7","NIP7","TERF2","VPS4A","MTSS2","ANKEF1","PCED1A","BTBD3","PCNA","NXT1","POLR3F","RIN2","PTPRA","DAP3","RHBG","KHDC4","ATP1A4","BCAN","ARHGEF11","RAB25","FCRL2","DCAF8","SYT11","IGHMBP2","ACY3","ALDH3B2","TESMIN","MMACHC","DPH2","TOE1","NASP","MUTYH","CTNNBL1","LPIN3","ZSWIM3","RBM38","VSTM2L","OSER1","SERINC3","PPP1R3D","DMGDH","BHMT2","AP3B1","ZBED3","PATJ","ANGPTL3","SLC14A2","FBXO44","CPLANE2","CASP9","DCTN4","PDE6A","ATP8A2","MTUS2","ZMYM5","USPL1","XPO4","TPTE2","POMP","CDK8","ALOX5AP","WASF3","CHRM3","MYH10","PEMT","SCO1","MPRIP","CHI3L1","PIK3C2B","DSTYK","CHIT1","SLC41A1","LGR6","TMCC2","DCLK1","CCNA1","COG6","SPART","EPSTI1","POSTN","RFXAP","TPT1","GPALPP1","STOML3","RFC3","STARD13","MORC4","RNF128","TBC1D8B","TCEAL4","FAM104A","SLC39A11","EPHB2","SRRM1","BTBD2","PRAM1","KMT5C","ZNF414","PDE6B","HSPBP1","CSNK1G2","SLF1","CNDP2","MACROD1","WDR74","RTN3","PLAAT4","PLAAT2","MYH11","FOPNL","MED10","PDZD2","MORC2","LARGE1","GSTT2B","SLC2A11","C1QTNF6","FAM83F","SEC14L4","GIMAP6","GIMAP4","ADCK2","MKRN1","AGAP3","KRBA1","ACTR3B","NTS","BTG1","LRRIQ1","C12orf29","ATP13A3","DYDC2","TMEM254","TMTC1","KRAS","IPO8","LARS1","SPINK5","IMPA1","LRRCC1","E2F5","CA1","CCDC59","SWAP70","ARNTL","LYVE1","AMPD3","SBF2","MICAL2","RRAS2","HSD17B4","ZFC3H1","SARAF","RNF122","DPF2","MEN1","ERG28","DGLUCY","UNC79","NUMB","CATSPERB","COX16","TTC9","MED6","EIF2S1","ADAM20","LOXL2","ELP3","ADAMDEC1","CTIF","MRO","MBD2","IER3IP1","MRPS36","CCNB1","CDK7","CD180","IRAK2","CAMK1","THUMPD3","VHL","BHLHE40","ARL8B","EDEM1","CHL1","MEIS2","DPH6","KATNBL1","EMC7","GNAT2","GSTM1","PRPF38B","TSPAN2","GSTM3","VAV3","PSRC1","HMGCS2","PTPN22","SORT1","WNT2B","PTGFRN","LAMTOR5","NOTCH2","TRIM45","CEPT1","CD101","VTCN1","AP4B1","NAPG","SPIRE1","PPHLN1","FKBP11","ARF3","TMEM106C","SLC38A2","YWHAQ","KIDINS220","GRHL1","ROCK2","RSAD2","LPIN1","CMPK2","IAH1","LDHA","SAA2","ANO3","IL6ST","FST","NAV1","CDC73","TIMM17A","CRB1","ERN2","RPS15A","NARS1","RELCH","FBH1","RBM17","IL2RA","ANKRD16","ECHDC3","IL15RA","CCNH","HRH4","TMEM241","KCTD1","CABLES1","DOCK2","EMP1","SOX5","RERG","KLRD1","KLRC1","PRH2","LRP4","MYBPC3","DDB2","USP26","RTL8C","RBMX2","STK26","PIWIL4","PUM1","SPOCD1","YARS1","PHC2","CDCA8","GNL2","AGO4","HOOK1","CYP2J2","BTF3L4","TUT4","PRPF38A","DSC2","DSG3","RNF138","ELP2","DSC3","DTNA","FHOD3","TPGS2","DAGLA","SLC43A3","TIMM10","CBLIF","DHX34","FADS2","TMEM258","TCN1","C5AR2","TMEM165","CLOCK","PDGFRA","GGACT","COL4A2","CLDN10","DZIP1","UBAC2","ARGLU1","BIVM","ERCC5","TPP2","POGLUT2","CARS2","ARHGAP32","STT3A","ADAMTS8","ETS1","SLC37A2","KLB","TMED7","APC","NREP","WDR36","OSTF1","RFK","UBQLN1","NAA35","C9orf40","ANXA1","CTSL","CEMIP2","AGTPBP1","GOLM1","FAM189A2","PSAT1","ISCA1","ADAM19","HAVCR2","CCNJL","TAOK3","USP30","SDS","MSI1","FBXO21","OASL","HRK","RNFT2","P2RX4","BICDL1","DTX1","TRAFD1","DMTF1","TMEM243","CCDC146","TMEM60","CD36","PNPLA8","HILPDA","RINT1","SRPK2","KCP","TES","MDFIC","MTO1","ANKRD6","KHDC1","CEP162","SYNCRIP","SNX14","NT5E","MRAP2","AKIRIN2","ORC3","LCA5","MAP3K7","PRR5L","LMO2","PHF21A","NAT10","EHF","ELF5","PRRG4","CAPRIN1","ATP5MC2","DNAJC14","CD63","PRPH","AVIL","GDF11","GLS2","ITGA7","TESPA1","FAM186B","RDH5","AGAP2","BLOC1S1","CDK4","PPP1R1A","TROAP","TSPAN31","TFCP2","COQ10A","FAIM2","PAN2","ESPL1","KRT7","ZC3H10","HNRNPA1","ACVR1B","OS9","KCNH3","LTV1","MAP7","CD164","AFG1L","NHSL1","AHI1","HEY2","PKIB","SMPD2","MICAL1","REPS1","STX11","TEC","PRADC1","SEMA4F","CCT7","RAB11FIP5","SMYD5","DYSF","CCDC142","KCNMB4","USP15","GNS","CPM","MDM2","KLHL36","BCO1","MPHOSPH6","CHST5","KIAA0513","DYNC1LI2","FBXL8","FHOD1","CCDC102A","SLC9A5","ZNF670-ZNF695","PCNX2","KCNK1","URB2","EGLN1","CAPN9","COG2","ABCB10","NTPCR","TAF5L","GLUL","STX6","RNASEL","DHX9","KIAA1614","CEP350","NPL","NIBAN1","PIGC","LAMC1","RC3H1","SP110","MRPL44","PAX3","DOCK10","TTLL4","USP37","HTR2B","ITM2C","SLC19A3","SERPINE2","DNAJB2","WNT10A","TMBIM1","CYP27A1","EIF4E2","ARMC9","CAB39","COX5B","REV1","TSGA10","MFSD9","TMEM127","EDAR","TGFBRAP1","GCC2","MRPS9","C2orf49","ANKRD36","EPC2","ARHGEF4","ISCU","ALDH1L2","STAB2","USP44","SCYL2","CKAP4","PLXNC1","APPL2","PWP1","DRAM1","WASHC4","SLC41A2","VILL","FLNB","NEK3","VPS36","RNASEH2B","CKAP2","TBC1D4","THSD1","BORA","LRCH1","SUCLA2","RCBTB1","MED4","PHF11","COG3","LMO7","SCEL","ITM2B","SPRY2","NUDT15","EDNRB","RCBTB2","LCP1","SETDB2","SCRN1","C7orf25","TNS3","SPDYE1","CHST12","IGF2BP3","GPNMB","RAPGEF5","RAC1","KDELR2","NUP42","IL6","ZDHHC4","AOAH","BZW2","DGKB","TBRG4","DDX56","HUS1","NACAD","DBNL","CCM2","MYO1G","TTYH3","CIDEB","TTC5","ZFHX2","MTHFS","ADAMTS7","ABHD17C","IREB2","ALPK3","TM6SF1","CIB2","CALCOCO2","RSAD1","NMT1","MYCBPAP","SRSF1","VEZF1","CHAD","TACO1","TEX2","DCAF7","LIMD2","BRIP1","KAT7","RTP4","ACTL6A","NDUFB5","MRPL47","TRA2B","SCN2A","TBR1","MARCHF7","ERMN","GALNT5","SCN7A","TANK","BLK","SKIL","EPRS1","HLX","VPS45","IL10","KCTD3","RPS6KC1","CBWD2","IL1RN","IL36A","IL36RN","SMPD4","WDR33","CCDC115","SAP130","BIN1","IMP4","HS6ST1","UGGT1","GYPC","STAM","GAD2","ABI1","YME1L1","DNAJC1","NIPSNAP3A","LRRC8A","CDK9","TXN","ODF2","ECPAS","TOR1B","C9orf78","SMC2","KLF4","TOR1A","RALGPS1","NIBAN2","ST6GALNAC4","DAB2IP","STXBP1","SLC2A8","ANGPTL2","CDK5RAP2","ZFP37","SLC31A2","SLC31A1","TLR4","ZNF189","ALDOB","STX17","PRPF4","FPGS","USP20","BAAT","ATP6V1G1","TEX10","GARNL3","MRPL50","DPM2","WDR38","TSTD2","GABBR2","HEMGN","PSMB7","TRMO","RABEPK","GOLGA1","XPA","NCBP1","ANP32B","PDCL","RPL35","CTSV","LMX1B","ARPC5L","ENPP2","DSCC1","DERL1","MYC","IL33","DMAC1","RANBP6","POLR1E","PLAA","IL11RA","UBAP2","APTX","RNF38","TLN1","SIT1","DMRT1","DNAJB5","SPAG8","DCTN3","CD72","TMEM8B","GRHPR","ALDH1B1","HINT2","ARHGEF39","IGFBPL1","DENND4C","RPS6","CNPY3","FOXP4","PPIL1","KLC4","KIF13A","ZSCAN9","PIM1","GMPR","CMTR1","TFAP2A","YIPF3","TMEM14B","TMEM63B","FRS3","TJAP1","KIAA0319","IRF4","SLC22A23","TUBB2A","LRRC1","GCM1","BPHL","RIPK1","UQCC2","HMGA1","TCF19","FLOT1","IER3","MDC1","PGBD1","ATAT1","TPMT","RNF144B","NRM","MTCH1","VARS2","TAF8","FAM8A1","C6orf52","FGFBP1","FGFBP2","CPEB2","FHDC1","TLR2","TTC29","MYO7A","FCHSD2","ARRB1","SLCO2B1","THAP12","ANKRD42","IL18BP","NUMA1","CCDC90B","SYTL2","RAB30","CREBZF","PRCP","NARS2","RNF121","MRPL15","TTPA","GGH","SLCO5A1","SULF1","TGS1","SDCBP","NEK1","DDX60","SORL1","TMPRSS4","BUD13","TRPC6","MMP7","CFAP300","DCUN1D5","YAP1","TRIM29","SLC37A4","BTG4","POU2F3","RDX","PPP2R1B","FDX1","C11orf1","FXYD6","ARHGAP20","MMP13","TMPRSS13","CASP1","CASP5","ALKBH8","MAP2K5","UNC13C","SQOR","CTDSPL2","SLTM","THBS1","MAPKBP1","NUSAP1","NDUFAF1","KIF23","ITGA11","KNL1","HAUS2","RTF1","PARP6","RPLP1","PAQR5","LRRC49","TUBGCP4","RMDN3","ITPKA","UACA","SMAD6","PLCB2","TMEM62","PAK6","ADAM10","DUOX1","SLC28A2","STRA6","CYP19A1","ZNF280D","SEMA6D","RSL24D1","SPTBN5","GCOM1","GCHFR","BCAR3","TTLL7","FNBP1L","KYAT3","GTF2B","RABGGTB","IFI44L","GIPC2","ARHGAP29","IFI44","SLC44A5","CLCA2","DBT","RTCA","IFT172","SELENOI","HADHB","ADCY3","PPM1B","PNPT1","DYNC2LI1","THUMPD2","CYP1B1","RAB1A","ACTR2","PREB","SLC5A6","ABCG5","PREPL","SLC3A1","FBXO11","SIX3","ATRAID","CENPO","LRPPRC","DTNB","ACTR1A","CYP2C9","MFSD13A","CYP2C8","MYOF","LOXL4","STAMBPL1","CH25H","ATAD1","BTBD16","KIF11","TACC2","DUSP5","CALHM2","ARL3","CEP55","KIF20B","ENTPD1","EXOC6","PLCE1","DBR1","DNAJC13","GPR87","ANXA7","FAM149B1","ASCC1","ZNF365","OIT3","ADAMTS14","RPS24","TET1","DNA2","MYPN","AOX1","ATIC","SMARCAL1","BARD1","STAT4","CARF","ASNSD1","METTL5","SSB","NAB1","PPIG","FASTKD1","MDH1B","HECW2","IDH1","OLA1","CIR1","ITPRID2","CHRNA1","FAM117B","WDR12","ABI2","ITGAV","SLC40A1","SLC35A5","SLC49A4","SENP7","COX17","PARP9","MNS1","USP8","SECISBP2L","TMOD3","SPPL2A","GLCE","SHF","APH1B","INTS14","PARP16","PPCDC","HCN4","SEMA7A","UBL7","ARHGAP24","FAM13A","HERC3","HERC6","HERC5","ZGRF1","AP1AR","COPS4","HNRNPD","PRKG2","RASGEF1B","SEC31A","FGF5","GPAT3","FGF2","BBS7","KIAA1109","BMPR1B","RAP1GDS1","LARP1B","PDE5A","PRDM5","TRPC3","NAAA","NUP54","CXCL9","BMP2K","G3BP2","SEPTIN11","FRAS1","SCARB2","CCNG2","CNOT6L","USO1","CDKL2","SHROOM3","ANXA3","PPA2","CENPE","GSTCD","INTS12","CASP6","LEF1","HADH","EGF","PAPSS1","SEC24B","C4orf17","PPP3CA","SLC39A8","MTTP","FBN2","MAPK8IP3","RGS3","GUCD1","RNF185","PARVG","B4GALNT3","ERP27","ETV6","GABARAPL1","KIF21A","CPNE8","YARS2","FGD4","ALG10","PIK3C2G","SINHCAF","AEBP2","ETFBKMT","ETNK1","ZCRB1","TMEM117","PRICKLE1","C1RL","NDUFA9","CLSTN3","KLRG1","VAMP1","TAPBPL","CD27","RBP5","PEX5","SLC38A4","AMIGO2","SCAF11","LLPH","LRIG3","MARCHF9","INHBE","GLIPR1","TPH2","PHLDA1","TMEM19","PTPRQ","DUSP6","POC1B","TMTC3","SNRPF","AMDHD1","NEDD1","SYCP3","GAS2L3","TMEM132B","SLC15A4","TDG","RITA1","SDSL","MMAB","GLTP","GIT2","TCHP","FAM222A","FOXN4","NUP58","MTMR6","SLC46A3","SLC7A1","LNX2","SUOX","CCDC65","TARBP2","RDH16","DHH","ACVRL1","GPR84","NABP2","N4BP2L1","SMARCC2","BRCA2","KANSL2","CERS5","MAP3K12","ITGB7","GALNT6","CSAD","LMBR1L","C12orf10","ESYT1","TMBIM6","ANKRD52","ZNF740","WDFY2","HNRNPA1L2","LPAR6","ESD","RB1","SBNO1","MORN3","SETD1B","VPS33A","VPS37B","RHOF","DENR","DIAPH3","SLAIN1","RBM26","SRRM4","MBNL2","RNF113B","ABHD13","RAB20","GRTP1","CUL4A","TTC6","CDH24","REM2","CBLN3","TSSK4","NOVA1","TMX1","FRMD6","PELI2","RTN1","ARMH4","SYT16","SLC38A6","NAA30","ADAM21","RDH12","DCAF5","RAB15","WDR89","ESR2","STON2","EFCAB11","GPR65","PTGR2","JDP2","AK7","FAM181A","SLC24A4","FBLN5","SERPINA10","CLBA1","WARS1","SLC25A47","WDR20","NIPA2","SLC12A6","DUOXA1","MFAP1","TCF12","SORD","SERF2","ZSCAN29","DUOXA2","DUOX2","LYSMD2","SLC27A2","FGF7","HDC","GCNT3","BNIP2","GTF2A2","SRP14","BAHD1","DISP2","CDAN1","TLE3","ANP32A","COMMD4","UBE2Q2","PSTPIP1","ETFA","BCL2A1","HMG20A","SCAPER","TSPAN3","WDR61","NCOA2","NEIL1","MAN2C1","DNAJA4","TLNRD1","TPM1","IGF1R","ARRDC4","PIF1","USP3","CYP11A1","BBS4","PML","ADAMTS17","LINS1","ULK3","PCSK6","CCDC33","CELF6","SCAMP2","CYP1A2","LMAN1L","HAPLN3","RHCG","POLG","RLBP1","FANCI","ABHD2","WDR93","TICRR","NTRK3","DET1","MFGE8","ZNF710","UNC45A","MCTP2","FURIN","IQGAP1","CRTC3","EFL1","SH3GL3","SEC11A","GLYR1","PMM2","SLC5A2","ITGAX","TGFB1I1","C16orf58","ARMC5","PARN","FTO","UQCRC2","CDR2","IGSF6","ARHGAP17","MYLK3","NKD1","DHX38","TXNL4B","MARVELD3","CHST4","ZFHX3","CLEC18B","CPNE2","NLRC5","KATNB1","KIFC3","NUDT7","GCSH","CMTM3","CMTM2","CDH11","NOL3","MAP1LC3B","MBTPS1","CDH13","ZCCHC14","MEAK7","ADAD2","OSGIN1","IRF8","RHOT2","RPL3L","ZSCAN32","RPS2","NDUFB10","PDPK1","TIGD7","DEF8","TCF25","GALNS","GAS8","MED9","NCOR1","COPS3","GID4","ZNF287","KSR1","UTP4","RANBP10","CTRL","GFOD2","NOB1","PRPSAP2","UNC45B","PCTP","OR4D1","TOM1L1","C17orf80","TOB1","VPS53","SGSM2","NPEPPS","SKAP1","LRRC46","SCRN2","SSH2","ARSG","ABCA8","G6PC3","CLTC","BCAS3","PTRH2","SS18","TAF4B","AFG3L2","PRELID3A","IMPA2","GNAL","SLC39A6","RPRD1A","C18orf21","GALNT1","ASXL3","MEP1B","GAREM1","ESCO1","OSBPL1A","GATA6","GREB1L","RMC1","PELP1","NPC1","SLC14A1","ARRB2","SLC13A5","ZMYND15","WRAP53","MINK1","SAT2","ASGR1","PIK3R5","TP53","CCDC40","ARHGDIA","TMC6","SLC16A3","CARD14","TTYH2","RAB40B","EIF4A3","CSNK1D","ANAPC11","TBCD","FN3KRP","NARF","RPTOR","FOXK2","TRIM65","CBX8","SECTM1","RNF157","CEP131","ZNF750","WDR45B","CBX4","RNF165","DYM","ELAC1","MBD1","SMAD4","TNFRSF11A","ZCCHC2","FBXO15","CBLN2","PMAIP1","P3H4","NT5C3B","RETREG3","ERBB2","GRB7","MIEN1","STAC2","IGFBP4","FKBP10","TXNL4A","CACNA1A","MISP3","SAMD1","BRD4","SLC39A3","NFIC","TPGS1","PLPP2","PRDM15","PFKL","FEM1A","VAV1","MVB12A","SH3GL1","DUS3L","DPP9","CCDC97","TMEM91","ZFP14","SIRT3","IFITM3","PGGHG","HUNK","COL6A1","IFNAR1","SOD1","COL6A2","SIK1","TRPM2","SCYL1","TMEM50B","APP","DOP1B","URB1","AKT1","IL19","EMP3","SAE1","NTN5","LMTK3","GEMIN7","CBLC","WTIP","ADAMTS10","RNPEPL1","CAPN10","MYO1F","ERVK3-1","NLRP12","CACNG8","ZNF787","TIMM29","CARM1","EVI5L","SLC47A1","PSMB6","SIGLEC10","ZNF473","FAM71E1","RPS11","RPL13A","CTU1","NOSIP","RCN3","ZNF614","SLC2A5","RERE","MMEL1","CFAP74","PRDM16","CELA2A","PADI3","FHAD1","PADI1","EPHA2","ARHGEF19","EFHD2","PEX14","PGD","MYOM3","SH3BGRL3","CNKSR1","RPL11","IL22RA1","ZNF593","C1orf216","KIAA0319L","EVA1B","DMRTA2","PLK4","MAP3K6","FCN3","GPN2","SYTL1","WDTC1","NBPF3","HSPG2","ITGB3BP","SERBP1","BCL10","CCN1","PRKACB","PIGK","TINAGL1","AZIN2","RPS8","KIF2C","PTPRF","BEST4","MOB3C","CYP4B1","TMEM61","LMO4","MTF2","SLC44A3","IGSF3","ZNF697","CTTNBP2NL","STRIP1","PSMA5","C1orf162","CD53","CELSR2","ITGA10","GPR161","ALDH9A1","ATP1B1","TIPRL","NME7","POGK","MPC2","CREG1","DCAF6","TBX19","UCK2","TMCO1","POU2F1","ILDR2","MGST3","ADCY10","COP1","NECTIN4","UFC1","PPOX","FCGR2A","NUF2","RGS5","SDHC","PFDN2","NR1I3","USP21","PRCC","FCRL5","RRNAD1","MRPL24","PIGM","ISG20L2","CRABP2","HDGF","ABL2","XPR1","RGS16","TOR1AIP1","HMCN1","RGL1","LYPLAL1","PRUNE1","RORC","TUFT1","SF3B4","ECM1","ZNF687","TARS2","CGN","SNX27","SETDB1","ADAMTSL4","MCL1","CTSK","RFX5","PI4KB","PIP5K1A","ANP32E","MINDY1","ANXA9","SELENBP1","CERS2","ENSA","SEMA6C","MRPL9","ARNT","POGZ","C1orf56","OAZ3","HORMAD1","GOLPH3L","GABPB2","SYT14","KCNH1","DTL","DYRK3","EIF2D","INTS7","VASH2","TAF1A","SMYD2","SUSD4","DUSP10","TP53BP2","ATP8B2","FLG2","CRNN","ADAM15","JTB","RAB13","S100A8","TPM3","NUP210L","SNAPIN","SLC27A3","S100A7","UBAP2L","SLC39A1","HAX1","CREB3L4","EFNA3","KCNN3","C1orf43","GATAD2B","ILF2","RIT1","INTS3","HCN3","FLG","C1orf131","GALNT2","TTC13","SCCPDH","LYST","MAP3K21","CEP170","ACP1","SNAP47","SRP9","NVL","SDE2","DEGS1","FBXO28","ARF1","CNIH4","ITPKB","GUK1","CDC42BPA","CNIH3","C1orf35","MBOAT2","PARP1","PSEN2","PYCR2","LBR","WNT9A","EPHX1","REN","SOX13","ETNK2","PPFIA4","PLEKHA6","PTPN7","SYT2","ARL8A","OSR1","GDF7","PDIA6","RHOB","ATP6V1C2","HNRNPLL","GALM","CAMKMT","ABCG8","EML4","CALM2","CHAC2","RPS27A","WDPCP","VPS54","ASXL2","ETAA1","SNRPG","ABHD1","MEIS1","CIAO1","ZNF514","SNRNP200","MRPS5","ANKRD53","TPRKB","NAT8","EXOC6B","SFXN5","TEX261","DQX1","DUSP11","ST6GAL2","NPHP1","MALL","THNSL2","RALB","TMEM177","NT5DC4","RABL2A","SLC20A1","FBLN7","ZC3H8","LIPT1","CNGA3","FAHD2B","AFF3","UBXN4","SPOPL","POLR2D","AMMECR1L","GALNT13","PKP4","SCN1A","SLC4A10","SCRN3","LNPK","ZNF385B","CDCA7","UBR3","PHOSPHO2","GULP1","FAM171B","HSPD1","CCDC150","METTL21A","UNC80","PTH2R","NBEAL1","KANSL1L","SPAG16","ABCA12","SUMF1","RHBDD1","ACKR3","TRPM8","HES6","ANKMY1","COPS7B","DIS3L2","FANCD2","TAMM41","VGLL4","RAB5A","RETREG2","CTDSP1","CNOT9","MARCHF4","STK11IP","GMPPA","GRIP2","EAF1","CNTN4","DYNC1LI1","RBMS3","GADL1","OSBPL10","POMGNT2","ACKR2","CSRNP1","SLC25A38","ITGA9","SLC22A14","GOLGA4","CTDSPL","IQSEC1","CAND2","RPL32","PTPRG","IL17RD","SHQ1","SLC25A26","UBA3","ARL6IP5","TMF1","LRIG1","LRTM1","LIMD1","ZNF660","NFKBIZ","COL8A1","NXPE3","MYH15","PHLDB2","ABHD10","TAGLN3","PLA1A","RABL3","ADPRH","IGSF11","ATG3","NR1I2","BOC","SRPRB","TMEM108","MED12L","EIF2A","ALDH1L1","OSBPL11","TRPC1","NCEH1","FAM86B2","LPP","TMEM44","RUBCN","AMT","TCTA","NICN1","DCAF1","MANF","STXBP5L","EAF2","ILDR1","TM4SF19","MUC4","SLIT2","EIF2B5","DGKQ","FIP1L1","SLC26A1","LYAR","CENPC","CORIN","ATP10D","OCIAD2","SLC10A6","SCD5","PLAC8","ENOPH1","TRMT10A","KLHL8","SNCA","PYURF","TBCK","CAMK2D","CISD2","DDIT4L","ANK2","TIFA","SPATA5","FABP2","CCNA2","METTL14","USP53","SETD7","NAF1","MARCHF1","RPS3A","RNF175","PDGFC","CBR4","GLRA3","CYP4V2","ROPN1L","NDUFS6","MARCHF6","CDH18","ADAMTS16","SRD5A1","MYO10","OTULINL","RPL37","SKP2","OSMR","UGT3A1","PLK2","GZMA","PIK3R1","LHFPL2","SSBP2","BHMT","ANKRD31","IQGAP2","RASA1","LIX1","GIN1","PPIP5K2","PAM","BDP1","GTF2H2","SLC30A5","BTF3","FBXL17","TNFAIP8","FEM1C","COMMD10","ATG12","MEGF10","YIPF5","ARHGAP26","CXCL14","LECT2","SLC25A48","DDX46","RNF145","GABRB2","FBXO38","SPINK7","PCYOX1L","GLRA1","TNIP1","G3BP1","ZNF300","N4BP3","NHP2","RMND5B","BOD1","CPLX2","TENM2","KCNMB1","FAM50B","MYLK4","FAM217A","TBC1D7","FARS2","GFOD1","CDKAL1","LRRTM2","ZMAT2","KLHL3","DCDC2","TRIM7","TRIM41","HIGD2A","FAM193B","PLA2G7","TNFRSF21","RNF44","MMUT","DOK3","ABT1","PPP1R18","DAAM2","PRIM2","MLIP","LGSN","FGD2","SCUBE3","ANO7","TCTE1","RPL7L1","NFKBIE","CYP39A1","TPBG","IRAK1BP1","PHIP","MMS22L","FAXC","PNRC1","PM20D2","RARS2","SCML4","TBC1D32","RNF217","ARHGAP18","ABRACL","SLC18B1","MTFR2","SLC2A12","SHPRH","AIG1","DYNLT1","TIAM2","TMEM181","PNLDC1","WTAP","ZMYM4","ARMT1","VWDE","GNA12","C7orf50","SDK1","CCZ1B","C7orf26","RBAK","CREB5","EGFR","CDCA5","IGFBP3","PURB","MDH2","POMZP3","NIPSNAP2","CCT6A","PSPH","ZNF92","ATXN7L1","TMEM168","ASB15","MAP11","SLC12A9","GIGYF1","TRIM4","MEPCE","TMEM209","AGBL3","ZC3HAV1L","TMEM140","TLK2","EPHA1","NOM1","CNPY1","NCAPG2","NLGN4X","SHROOM2","RAB19","LUC7L2","DENND2A","CLTRN","SH3KBP1","TMEM47","LANCL3","SYTL5","CASK","KDM6A","SPIN2A","MSN","CCNB3","HDAC8","SLC16A2","DIPK2B","ZNF157","ZNF182","CHST7","KRBOX4","NDUFB11","ZNF41","ZMYM3","TAF1","GPR174","NONO","CCDC120","LPAR4","EBP","OGT","SNX12","ITGB1BP2","IL2RG","GCNA","ZNF711","DIAPH2","NXF3","PRPS1","RADX","FRMPD3","DOCK11","IGSF1","GPC3","RBMX","MCPH1","MFHAS1","FBXO25","NSDHL","ZNF185","CETN2","RPL10","CSGALNACT1","ATP6V1B2","CCDC25","HMBOX1","GNRH1","BIN3","DOK2","SLC25A37","CHMP7","DOCK5","STAR","PLPBP","ERLIN2","SNTG1","ST18","RGS20","TACC1","GOLGA7","PLPP5","GINS4","NSD3","DNAJC5B","ADHFE1","MRPS28","PMP2","LACTB2","PRDM14","TERF1","RPL7","SLC26A7","ATP6V0D2","SYBU","DPYS","MTDH","LRP12","EBAG9","RSPO2","POLR2K","MAL2","EIF3H","UTP23","NDUFB9","TATDN1","FAM83A","GSDMC","FAM135B","ZNF7","ARHGAP39","SLC39A4","NAPRT","VLDLR","AK3","UHRF2","NFIB","PLIN2","HAUS6","CDKN2B","CDKN2A","C9orf72","IFNK","ZCCHC7","FBXO10","SIGMAR1","CBWD5","CEP78","NTRK2","IDNK","SHC3","AUH","MFSD14B","AOPEP","ZNF462","INIP","UGCG","SNX30","STOM","GSN","MRRF","NR6A1","CRB2","ALAD","ASTN2","WDR31","POLE3","SURF4","GBGT1","SURF1","SURF2","SURF6","MED22","REXO4","RPL7A","GTF3C5","ASB6","PTGES2","NTMT1","CIZ1","SLC25A25","SH3GLB2","MIGA2","PTGES","LCN2","LRSAM1","HMCN2","GPR107","PAXX","INPP5E","SEC16A","DPH7","NOTCH1","CACNA1B","NACC2","PROSER2","USP6NL","COMMD3","MSRB2","PDSS1","FAM171A1","MINDY3","TMEM236","RSU1","ST8SIA6","PARD3","ZEB1","FAM13C","NRBF2","A1CF","RGR","POLR3A","HERC4","LRMDA","CAMK2G","ADIRF","GLUD1","HTR7","RPP30","FRA10AC1","ADD3","DNAJB12","EIF4EBP2","NPFFR1","PLEKHS1","TCF7L2","MKI67","CYP17A1","FUOM","LRRC27","MTG1","PAOX","GSTO1","TAF5","PPRC1","ITPRIP","CNNM2","PDCD11","ADAM12","RGS10","BTBD10","ADM","GAS2","SLC5A12","LIN7C","IMMP1L","SAA4","PGAP2","TUT1","SCGB1A1","SYT8","ZNF214","ZNF215","HSD17B12","APIP","PAMR1","DGKZ","EIF3M","TNKS1BP1","GLYAT","SERPING1","SSRP1","SLC43A1","PTPRJ","C11orf49","ARFGAP2","CELF1","HIKESHI","CCDC81","SESN3","ENDOD1","CCDC82","KLHL35","TENM4","SERPINH1","CAPN5","INTS4","PAK1","RPS3","ZC3H12C","TTC12","NCAM1","DRD2","C11orf52","HTR3B","NPAT","ATM","AASDHPPT","GLB1L2","SLX4IP","LAMTOR1","P4HA3","GRIK4","ST14","HYOU1","ADAM33","KAT14","TKFC","MTA2","TMEM138","FADS1","TMC2","ROM1","EML3","INCENP","PLCH2","CPSF7","MS4A2","B3GAT3","EI24","CCDC15","CHEK1","FEZ1","ESAM","KIRREL3","MPZL2","SCN2B","SIDT2","TMEM25","TAGLN","COMMD7","C20orf144","DSN1","SOGA1","CNBD2","CDH22","LSM14B","YTHDF1","CABLES2","LTO1","SLC22A9","TRPT1","NUDT22","FERMT3","PLCB3","MRPL49","CDC42EP2","FAU","TM7SF2","VPS51","TBX6","PPP4C","ALDOA","TLCD3B","DOC2A","HIRIP3","TAOK2","TMEM219","HMGA2","MMP3","CNKSR2","CNTN5","KLRF1","CLEC1A","MKX","MPP7","ITGB1","PCDH15","CTF1","CWC15","FCGR1A","ARID5B","CDH8","DCUN1D2","TMCO3","TMEM218","TIRAP","EEF1AKMT1","LATS2","SAP18","ADGRL3","KIAA1328","FAM124A","MIA2","HNMT","LYPD1","LYPD6B","PDCD4","ADRA2A","GPM6A","WDR17","SPATA4","VEGFC","CCDC102B","CD226","CNDP1","FSIP1","DLG2","CCDC83","RGS18","PRSS23","MTMR12","PPP1R1C","C11orf53","CCT5","ATPSCKMT","DOCK1","DIXDC1","DLAT","PIH1D2","NKAPD1","TIMM8B","IL18","PTS","PIP4K2A","C2orf50","FREM2","FOXO1","CRIM1","SEC24D","ABCB9","RILPL2","DHX37","UBC","ITPR1","PRSS53","SLC7A11","NOCT","ENKUR","CACNA2D4","DCP1B","CACNA1C","THRB","NGLY1","OXSM","UEVLD","TMEM86A","C12orf45","TMEM263","BTBD11","UBE3B","ANK3","IPMK","RAD9B","PLBD2","DLG5","MAT1A","SLC2A13","GXYLT1","TWF1","DIP2C","EIF4E","MAGI1","TEX30","CSNK1G3","SRFBP1","AKAP6","NPAS3","FAM177A1","MBIP","MIPOL1","EXT2","TMEM18","KCTD14","NDUFC2","ME3","ADAMTS12","NUBPL","NEK7","FER","VIPAS39","ANKRD50","UPF2","CDC123","SCLT1","C4orf33","FRMD4A","PTPRO","EPS8","ACAD8","THYN1","VPS26B","NCAPD3","VTI1A","QDPR","FAM160B1","ANO4","TEX9","QTRT2","MMAA","ZNF827","NR3C2","AKR1C2","VENTX","ADAM8","ITIH2","KIN","PIGF","ANKAR","INPP1","MFSD6","RNF144A","ASAP2","ADAM17","FLI1","KCNJ1","TMEM45B","WWC2","CENPU","ACSL1","SLC25A4","AMN1","BICD1","SAV1","CCDC122","SERP2","NBAS","ZNF385D","GUF1","SLC35F4","GABRA2","SACS","CCDC175","CENPJ","FBXO4","TMEM267","CCL28","PARP8","CACUL1","DST","BEND6","TIAL1","BAG3","GLT1D1","SCHIP1","MCHR2","KCNE4","AP1S3","RABGAP1L","CCDC74B","TLCD4","MZT2B","TUBA3E","FAM168B","PTPN14","MGAT5","TMEM163","GPATCH11","HSPB8","GEMIN6","TMEM178A","OBI1","CYSLTR2","ARL11","RIT2","SETBP1","ARL14EP","EPG5","PSTPIP2","ATP5F1A","HAUS1","C18orf25","SPC25","PDK1","PDE3B","TCF7L1","TGOLN2","SH2D6","UHMK1","ATG10","POC5","SPOCK1","FAM151B","TADA1","CWF19L2","JMY","HOMER1","XRCC4","BOLL","ZNF547","ZNF773","ZNF776","ZNF256","SUV39H2","DCLRE1C","OLAH","RPP38","NMT2","ZSCAN1","ZNF837","USP12","CCDC50","CAMK4","TRIM36","ZFP36L2","PAN3","PLEKHH2","PFKM","TMEM123","GRIA4","IGSF10","SPEF2","SPARCL1","MEPE","MBNL1","CAPSL","NADK2","GPD1L","GJA1","CCNO","SLC30A6","PELO","RASGRP3","SAR1B","CATSPER3","GPR180","TCTEX1D1","WDR78","ANKRD22","FARP1","IFIT5","SLC16A12","PANK1","PRDM8","BMP3","HNRNPDL","HHEX","UTRN","GRM1","PTPRK","GGPS1","ZNF117","LMNTD1","MARVELD2","RAD17","MED21","PLOD2","NRSN1","JAKMIP1","ZIC1","ADGRA3","CPB1","SREK1IP1","CWC27","MR1","SRP19","CENPH","CDYL","CARHSP1","TEKT5","BANK1","TXNDC11","DAB2","ACOXL","BCL2L11","ANAPC1","CAST","SCOC","CLGN","CETN3","SMARCA5","SYCP2L","BMP6","RASSF3","HNRNPU","RANBP2","AHCTF1","MERTK","TMEM87B","NR4A2","CCDC148","PLA2R1","RBMS1","SCN3A","CD96","SLC25A27","ADGRF1","FAM49B","ASAP1","TRAPPC8","FAM81B","INO80C","LPCAT1","PLEKHG4B","NMRAL1","UBALD1","C16orf89","TMEM251","ING1","SPACA7","ADPRHL1","CMTM7","FBXL2","UBP1","RMND5A","CD8A","RPIA","LURAP1L","CNKSR3","GTF2E1","CFDP1","ZDHHC7","FAM92B","C7orf31","TMPRSS11D","JAZF1","CMIP","SPHKAP","KCNJ16","PID1","TRIP12","FBXO36","CEBPG","KCTD15","ZNF599","MCOLN2","LGI4","DDAH1","SREK1","CHD1","ANKFN1","DGKE","HS2ST1","MSI2","CACNA2D1","ZUP1","GDPD1","NUS1","SEMA3D","PPP2R5E","GRAP","AK5","CABYR","IMPACT","ANKRD29","SDHAF4","CHST9","THY1","DNAAF1","C16orf74","TBCEL","JPH3","ANKH","OTULIN","UBASH3B","ROBO3","TBRG1","RETREG1","CDH12","TOMM70","ABI3BP","ANGPT1","PITPNC1","CC2D1B","CERS3","PRKCA","LRRK1","CEP112","GAL3ST2","ABCA9","ABCA6","ABCA10","ABCA5","ENPP3","C4orf19","UCHL1","MIA3","DISP1","TNIK","FAM167A","NEIL2","PGM5","WNT3A","OBSCN","LONRF1","TRIM11","ENAH","CCSAP","ASZ1","SH3RF1","GBP5","BUB3","CCDC173","MMP21","DIPK1A","ATP5MC3","CNTNAP3B","SRSF12","PDLIM3","SORBS2","ELOC","LY96","CXADR","BTG3","C21orf91","CHODL","NCAM2","L3MBTL4","PDE1C","RABGEF1","MRPL39","JAM2","ATP5PF","GABPA","ADAMTS1","TSEN2","SLFN13","XPC","CCDC174","FGD5","FLCN","DPH3","OXNAD1","PLCL2","CXXC1","SKA1","PPP4R1","APCDD1","PIEZO2","MPPE1","USP43","RAB6B","EME1","EPHB1","ACSS1","ANKRD40","ZNF18","VOPP1","SEPTIN14","APOOL","CYP2U1","RSPH10B","FBXL18","CNTNAP5","PROM2","AK9","KLF10","PTPRN2","AZIN1","ATP6V1C1","PIP4P2","OTUD6B","CDK19","GTF3C6","TTC39B","AGPAT5","MMS19","PI4K2A","MARVELD1","ZFYVE27","GOLGA7B","GPR78","TRMT44","SLC25A28","HSPA13","SAMSN1","USP25","GRAMD2B","ZCCHC10","C16orf87","MOV10","RHOC","PPM1J","DBI","SLC16A1","HEATR3","NIFK","OXA1L","SLC7A7","LARP1","CNOT8","GRIA1","LRGUK","SETD9","MIER3","NUP205","ZKSCAN2","C9orf85","PIK3AP1","RBM45","TTN","VSIG4","PDIA4","KDM8","PDZD9","OTOA","KCTD18","FAM126B","FLACC1","C2CD6","TMEM237","FZD7","SPAG17","DEPTOR","FMN2","RNF20","PPARGC1B","ELMO1","SLC26A2","LSM11","MED7","SAXO1","RRAGA","SLC24A2","PXYLP1","RASA2","RMND1","SLA","TMBIM4","VBP1","RAB39B","CLIC2","AFF2","MICU3","GRIP1","VPS37A","PSD3","CARNMT1","MCU","ELMSAN1","CFAP70","GNA14","FAM161B","GNAQ","GPR61","ADK","KCNMA1","BATF","DCK","ADAMTS3","ALX3","DPY19L4","NDUFAF6","DRAM2","C8orf37","PPEF2","CFAP161","ADAMTSL3","ART3","WHAMM","CXCL13","N6AMT1","RWDD2B","USP16","CCT8","MAP3K7CL","NAA11","BACH1","CLDN8","TIAM1","SCAF4","RPGR","CDK20","PCGF6","ANKRD9","SFR1","SFXN2","ATP5MPL","FUT6","TDRD9","FGF18","PCDH1","SH3RF2","UQCRB","MTERF3","PTDSS1","PPP2R2B","RPL30","KCNS2","FAM122C","SUPV3L1","FAM122B","EEF1A1","FBXO43","HK1","TYSND1","PHF6","CD109","NODAL","UBE2L6","ZDHHC5","MED19","ZFAND3","NPTN","KAT6B","SAMD8","RAB11FIP1","UTP14A","AIFM1","MAPK13","BAG4","MS4A1","TBC1D31","WDYHV1","ATAD2","FBXO32","NSMCE2","ZNF689","PRR14","FBRS","FRRS1","PHKG2","MFSD14A","SASS6","ITGAD","ZIC3","MALSU1","VPS8","GALK2","LHFPL4","B3GNT7","MPV17L","BUB1B","PDE6D","EIF4A2","BRPF1","RPUSD3","TATDN2","GHRL","SEC13","EXOG","NTAN1","NMNAT2","ZFYVE9","ATP2B2","SLC6A1","SMG1","FCHO2","RBPMS","TMEM171","TIMP4","SYN2","NRG1","ODR4","CPT2","NECAP2","LRP8","PAXIP1","STEAP2","SSBP3","CLDN12","MMP14","FZD1","GATAD1","SUSD3","TMED6","CLEC18A","DHRS4","C1orf158","CLEC18C","DDX19B","ST3GAL2","FCSK","IL34","DHRS1","CACNA1D","ARSL","KIT","HYDIN","AASDH","ZNF19","CACNA2D3","RNF111","CCNB2","FAM81A","MYO1E","APPL1","AFAP1L1","TSC22D3","VPS26C","DYRK1A","KCNJ6","KCNJ15","ETS2","TSPAN18","LCA5L","SLC35B2","TMEM164","MX1","CREB3L1","C2CD2","TAB3","SLC38A10","C9orf43","PALM2AKAP2","ZNF618","DGKI","TMEM268","SVOPL","SNX22","UBN2","BRAF","SLC34A2","ACAN","PSMG3","CABP1","WDR19","SLC37A3","AP3S2","FMNL2","GAREM2","SPPL3","DPYSL5","DRC1","RAB28","PRXL2B","TNFRSF14","PANK4","MEGF11","C12orf43","PEX10","RER1","RADIL","SKI","WIPI2","LDLRAP1","AGAP1","KRTCAP3","ANKRD61","PAFAH2","SLC30A2","BABAM2","WDR66","MRPL17","DUSP2","GRHL3","UBXN11","NLRP14","PTPDC1","GALNT14","NCK1","HPD","RHPN1","TPRG1L","LRRC43","PRXL2C","XDH","XKR8","CNNM4","EYA3","DZIP1L","FANCC","MRAS","WASF2","ABHD3","ESYT3","FAIM","TENT5B","CLSTN2","COLEC12","RNF207","CUL4B","GPR153","SLC13A3","GPRASP2","RHBDL2","AUTS2","SHROOM4","H2BC5","H4C8","MITD1","EIF5B","RIBC1","TMSB15B","CATIP","CNOT11","KCNB1","TSPAN33","NRG2","AHCYL2","B4GALT5","CD1D","SPATA2","CD1C","FAM86C1","DNAH3","CD1E","NCF1","TSR2","PPP1R9A","ZC3H18","ZFAND2B","GDPD5","DYNC1I1","TMED4","PPP1R15B","COPG2","EMSY","GPAT4","PKD1L1","ZSCAN12","TAGLN2","ELK4","SLAMF8","SLC45A3","DUSP23","RNF166","NBL1","HTR6","F11R","USF1","SPATA2L","NIT1","DEDD","ZNF276","NPM2","EDA","VWA5B1","CDA","PINK1","B4GALT3","DMTN","ADAMTS4","FAM160B2","NDUFS2","FCER1G","APOA2","TOMM40L","CCAR2","WNT9B","CACHD1","CDC42SE2","RAPGEF6","EPB41","MIS18A","ALG8","FBXW5","CFAP298","SYNJ1","PAXBP1","IFNAR2","MRPL10","IFNGR2","GART","SON","DONSON","SV2A","LAD1","STC1","TNNI1","CSRP1","C1QC","ATP5MC1","RCAN1","UBE2Z","CIART","SNF8","CLIC6","CCDC24","RUNX1","IGF2BP1","CBR1","CBR3",null,"GJD2","MORC3","CHAF1B","SIM2","HLCS","GOLGA6A","SCUBE1","ARHGAP27","ADPGK","PTMS","PLA2G4D","PADI4","ADIPOR1","CYB5R1","PSMD4","ATP13A2","M1AP","PSMB4","BTG2","HK2","C1R","CELF3","ALDH4A1","STARD9","THEM4","TCHH","UBR1","AMFR","MED8","RGL4","RSPRY1","CCDC17","GPBP1L1","NAE1","TMEM69","ADGRG5","DRC7","ACE","TEPP","EFCAB14","SPON2","CHCHD6","CTBP1","LRRC36","TPPP3","ZDHHC1","ATP6V0D1","ZFYVE28","CARMIL2","C16orf86","PIP","FAM131B","RGS12","PSKH1","ZYX","ABR","LYPD5","CCDC117","ZNF230","CCDC107","ZNF222","NPR2","ZNF221","ZNF233","ZNF235","GNE","TNFRSF13C","OR3A3","ARHGAP35","PTGIR","CALM3","DFFA","CCDC28B","IQCC","TMEM234","BSDC1","ZBTB8A","ATAD3B","SSU72","UBE2J2","ZNF362","FNDC5","CPAMD8","NR2F6","ANKLE1","CCDC58","VMA21","KALRN","ABCG1","TFF3","TFF1","TMPRSS3","UBASH3A","RSPH1","SLC37A1","PDE9A","WDR4","NDUFV3","PKNOX1","CBS","U2AF1","HSF2BP","RRP1B","PDXK","G6PD","CSTB","RRP1","AGPAT3","TRAPPC10","GAB3","GATD3A","ICOSLG","AIRE","CFAP410","ZNF66","LRRC3","ITGB2","FAM207A","RALGDS","SPATC1L","LSS","VAV2","MCM3AP","C21orf58","PCNT","DIP2A","S100B","PRMT2","ZNF208","ADAMTS13","CACFD1","SLC2A6","ZNF761","C9orf116","ZNF714","GPSM1","C19orf47","CFAP157","TOR2A","ST6GALNAC6","SHKBP1","RDH13","ZER1","ZDHHC12","PKN3","SPTBN4","COX6B2","TMEM190","PLPP7","TAOK1","MED27","DEDD2","SIK3","MPZL3","JAML","NEK8","TLCD1","PCSK7","SAFB","CD3G","S100A1","CHTOP","CXCR5","ZBTB7B","FLAD1","SHC1","VPS11","NLRX1","ADAR","IL6R","UBE2Q1","CHRNB2","CRTC2","ANO10","FDPS","RUSC1","FAM189B","PAQR6","PMF1","SLC25A44","LMNA","CCR5","NBEAL2","CCDC12","UBQLN4","MYL3","PPP1R35","GPATCH4","LRRC71","FCRL3","AZGP1","FGFR4","NACC1","CYP11B1","HK3","LY6K","IER2","ZNF394","CPSF4","LY6E","VPS28","TONSL","PWWP3A","RECQL4","LRRC14","ZNF333","PPP1R16A","ORAI2","ALKBH4","SH2B2","MRNIP","SQSTM1","MGAT4B","RPL8","MAML1","LRWD1","FBXL13","NAPEPLD","SCGB3A1","PSMC2","CELF5","MFSD12","USP41","YDJC","DVL3","AP2M1","ABCF3","PCYT1A","FBXO27","DMKN","U2AF1L4","BDH1","NPHS1","THAP8","COX7A1","ZNF382","LRRC56","PLXDC1","PGAP3","IKZF3","GRIN2C","FDXR","SAP30BP","ACOX1","PRPSAP1","CYGB","SRSF2","ZNF577","TMEM143","ALDH16A1","DCD","ITGA5","SIGLEC11","ZNF385A","MPP3","IZUMO2","NAGS","LSM12","ASB16","EMC10","JOSD2","SHANK1","FAM171A2","DBF4B","PLCD3","FMNL3","AQP5","RACGAP1","LARP4","GRASP","RAVER1","SPC24","IP6K3","LEMD2","ALOX15","ZNF653","MED11","CXCL16","SCIMP","TNFSF13","SENP3","FGF11","EIF4A1","RPL26","POLR3K","SNRNP25","WDR90","JMJD8","CCDC78",null,"SPSB3","HS3ST6","TEDC2","CCNF","TBC1D24","AMDHD2","BICDL2","PAQR4","FLYWCH2","ZG16B","ZNF75A","ADCY9","SHANK2","CLPB","NEU3","CYB561A3","PPP1R32","ASRGL1","UBXN1","LBHD1","TTC9C","TAF6L","NXF1","STX5","SLC25A45","RPL29","ITIH3","SYVN1","ZFPL1","RPS6KA4","LRP5","TPCN2","CYP4A22","PDZK1IP1","TAL1","CMPK1","BEND5","COA7","ZYG11B","CZIB","MAGOH","ACOT11","FAM151A","PARS2","LEXM","BSND","USP24","PLPP3","NOL9","PRKAA2","KLHL21","ZSWIM5","GMEB1","SLC45A1","SELENON","AK4","JAK1","RAVER2","CTRC","LZIC","RBP7","KNCN","FBLIM1","SLC25A34","AKR7A3","DRAXIN","PDPN","LRRC38","DHRS3","MATN1","LAPTM5","SDC3","PEF1","SYNC","RBBP4","KIAA1522","TSSK3","TMCO4","UBXN10","CAMK2N1","ALPL","WNT4","TTLL10","SCNN1D","MXRA8","FAAP20","MEGF6","CCDC27","IL23R","C1orf87","NFIA","OMA1","MYSM1","TM2D1","USP1","FUBP1","NEXN","DNAJB4","ADGRL4","LRRIQ3","TYW3","SNX7","B3GALT2","FAM102B","HENMT1","AKNAD1","C1orf52","WDR63","GBP2","ATXN7L2","GBP4","ZNF326","GFI1","AGL","EXTL2","SLC30A7","ZNF281","ARPC5","NLRP3","ZNF496","TRIM58","IGSF8","DDR2","PEA15","PEX19","NCSTN","VANGL2","SLAMF6","OLFML2B","FCGR3B","SLC9C2","KLHDC9","C1orf74","FLVCR1","FAM71A","ATF3","RBM15","DENND2D","AXDND1","IER5","SNED1","BPNT1","SPATA17","C1orf115","BROX","NBPF20","ACP6","KIF26B","TFB2M","CNST","PPP1R21","KLHDC8A","OXER1","HAAO","B3GALNT2","MAPKAPK2","FCMR","PIGR","CAPN2","MRPL55","WDR26","REL","PUS10","PEX13","KIAA1841","TRIM17","RFTN2","DISC1","CAPN13","MEMO1","DPY30","TYW5","MAIP1","SLC66A3","ARL5A","LRATD1","KCNJ3","CLHC1","DUSP19","CFAP36","NUP35","CCDC138","FBXO41","ACTG2","WDCP","SMC6","VSNL1","CCDC74A","H3-3A","ANKRD30BL","COQ8A","SLC16A14","TEKT4","SGCB","SPATA18","NOSTRIN","CFAP221","SGPP2","INHBB","BBS5","SMARCAD1","HPGDS","PDLIM5","STPG2","NEURL3","RPRD2","ANKRD23","CTSS","MSX1","PACRGL","BNIPL","C1QTNF7","TNFAIP8L2","LYSMD1","SCNM1","TMOD4","VPS72","ERCC3","RNF149","IWS1","BOLA3","CDC42EP3","S100A11","SPRR3","DHX57","SPRR2D","ARHGAP25","S100A9","S100A12","TGFA","CCNYL1","FZD5","DCAF16","C1orf189","GNPDA2","ALPP","PAQR3","NIPAL1","ANTXR2","HELQ","MRPS18C","CGGBP1","ABRAXAS1","GPR155","DAPL1","PMVK","PBXIP1","CLDN1","PYGO2","HIPK1","DCST2","DCST1","COL6A3","INAVA","YY1AP1","KBTBD8","EOGT","LMOD3","NAXE","POGLUT1","SLC22A15","ATP1A1","SLC15A2","EIF4E3","PROK2","TEX55","LRRC58","FSTL1","LMOD1","ELF3","TMEM183A","TMEM169","IGFBP7","TRIM46","KRTCAP2","CXCR1","ARPC2","CCT3","TMEM79","SSR2","RNF25","STK36","NEK10","CCDC141","CIP2A","EOMES","CWC22","AZI2","TGFBR2","ANKZF1","HDAC11","GLB1L","STT3B","CHCHD4","NFASC","FCRL1","SGO2","SERPINI1","CLASP2","SUCLG1","NUAK2","PRKCI","MNDA","PYHIN1","IFI16","AIM2","EFHB","EIF5A2","RPL22L1","PPM1L","ICA1L","CTLA4","ICOS","RYBP","PPP4R2","CD200R1","GTPBP8","NEPRO","SPICE1","CCDC191","CADPS","CDS1","WDFY3","COX18","PTPN13","SYNPR","C3orf49","C4orf36","THOC7","ATXN7","PSMD6","PRICKLE2","ADAMTS9","PPM1K","ERICH6","GMPS","TIPARP","CCNL1","PTX3","HESX1","SLMAP","RPL9","SMIM14","RPP14","ABHD6","DNASE1L3","C3orf67","RBM47","APBB2","IL17RE","IL17RC","CRELD1","PRRT3","PCOLCE2","U2SURP","MTMR14","TTC14","CXCL3","CXCL5","MTHFD2L","CXCL1","RCHY1","PLSCR2","CCDC158","CPA3","GYG1","HPS3","TOPBP1","RYK","SNRK","TCF23","ZNF513","SLC4A1AP","PLB1","SPDYA","KIAA1143","KIF15","TGM4","WDR43","ZDHHC3","CDCP1","SLC6A20","LZTFL1","FYCO1","CCR1","LRRC2","ELP6","FBXO40","DTX3L","ZNF148","NMNAT3","SMIM12","ZMYM6","TPRA1","YEATS2","ZC3H12A","MEAF6","SNIP1","DNALI1","POLR2H","KLF15","CFAP100","LIPH","TMEM41A","RPN1","SENP2","IFT122","RFC4","RPL39L","BAP1","TKT","PRKCD","RFT1","SFMBT1","GNL3","PBRM1","UVSSA","TASOR","ARHGEF3","SLBP","LRPAP1","ZDHHC19","SLC51A","UBXN7","RNF168","PIGX","MELTF","S100P","ABLIM2","EXO5","CLDN19","C1orf50","ERMAP","ZNF691","AIMP1","SGMS2","METAP1","DNAJB14","H2AZ1","EMCN","SLC9B1","SLC9B2","BDH2","PGRMC2","CDC25A","ZNF589","FBXW12","PLXNB1","CCDC51","ATRIP","SHISA5","SPRY1","BSN","APEH","INTU","RNF123","HSPA4L","MFSD8","ABHD18","MON1A","MST1R","RAD54L2","TEX264","GRM2","DUSP7","POC1A","PPM1M","WDR82","C4orf3","PRSS12","NDST3","HMGB2","SAP30","SCRG1","MAD2L1","ANXA5","MAP9","GUCY1A1","FBXO8","CEP44","HPGD","TMEM144","GASK1B","NAA15","IL15","FAM160A1","ARFIP1","ICE1","HHIP","ANAPC10","ABCE1","OTUD4","LSM6","TMEM184C","PRMT9","ITGA2","MOCS2","EDIL3","TMEM161B","ELOVL7","NDUFAF2","ZNF474","LMBRD2","RANBP3L","NIPBL","RNF180","ADGRV1","SLC25A46","STARD4","PGGT1B","F2RL2","CCDC112","ANKRD33B","CMBL","C5orf63","PRRC1","F2RL1","AGGF1","WDR41","PRDM9","NDUFS4","SCGB3A2","SPINK1","GRPEL2","CDC20B","ARSK","RHOBTB3","GPX8","TIGD6","SERINC5","ENPP6","CAGE1","CASP3","PRIMPOL","ERAP1","ERAP2","CMYA5","EGFLAM","CFAP97","RICTOR","TENT2","EBF1","ANKRA2","UBLCP1","UTP15","TLR3","KLKB1","NSA2","GFM2","CCDC127","FOXQ1","ADGRF2","ACSL6","SEPTIN8","SHROOM1","GDF9","UQCRQ","LEAP2","GJB7","SLC35A1","GRIK2","CGAS","FABP7","TXLNB","CITED2","CREBRF","DCBLD1","SFXN1","SAMD3","TMEM200A","PDSS2","STXBP5","IL31RA","RAET1E","DAGLB","KIAA0895","STK17A","TRA2A","GALNT10","SAP30L","RPS14","HCN1","MYOZ3","COG5","BMT2","GPR85","SLU7","RP9","PTTG1","CAMLG","BMPER","RELL2","KCNK5","KIF6","ZNF12","SLC29A4","STEAP1","CDCA7L","SP8","MIOS","KIAA1324L","USP49","SYTL3","IQUB","HEY1","ZNF704","FABP5","SHH","TAGAP","COL1A2","CHMP4C","SLC13A4","BRI3","LMTK2","CTSB","DLC1","ADCY1","C7orf57","HNF4G","PEX2","RAD21","SLC30A8","MED30","TNFRSF11B","SBSPON","PHKG1","KCNV1","CSMD3","SPIDR","ORC5","DNAAF5","OSGIN2","DEFB1","SUN1","OXR1","GPR146","GPER1","TMEM184A","NOS3","MICALL2","CA3","INTS1","CDK5","SLC4A2","FASTK","TMUB1","FMC1","PHAX","ALDH7A1","FOXK1","COX6C","YWHAZ","BAALC","FZD6","SLC25A32","DCAF13","TP53INP1","INTS8","VIRMA","GEM","PDP1","TMEM67","WASHC5","RPP25L","FAM219A","C9orf24","SNAPC3","MYORG","NUDT2","TMEM65","PSIP1","CCDC171","UBAP1","SYK","NIPSNAP3B","ABCA1","NFIL3","LETM2","METTL2B","FXN","ZMAT4","TMEM71","MAMDC2","C8orf34","TMC1","ALDH1A1","KDM1B","HGSNAT","RASEF","GKAP1","KIF27","C9orf64","HNRNPK","SVEP1","TRPV6","ANKS6","FBP1","PGAP4","ZHX1","CFAP47","CYBB","DYNLT3","METTL27","MID1IP1","SHOC1","CXorf58","KIAA1958","PTCHD1","RNF183","PCDH19","PIGA","STRBP","CLDN3","GAPVD1","CARD19","WNK2","ATP7A","ZNF367","NLGN4Y","HDX","NDUFB6","AQP7","NOL6","AQP3","TRMT10B","VCP","PIGO","STOML2","BRWD3","SLITRK5","MELK","ARMC3","OTUD1","ARHGAP12","FAT3","DEUP1","HECTD2","FBXO33","INTS6L","LRRC18","SPTSSA","WRN","MARCHF8","TSHR","CFL2","SUGT1","GTF2A1","ZCCHC24","PGM2L1","PHYHIPL","SLC16A9","INPPL1","GJB2","CRYL1","REEP3","HEPACAM","SKA3","MICU2","DDIAS","PCF11","LRR1","RPL36AL","DNAAF2","DEPP1","ZNF22","KLHDC2","EML5","NEMF","RPUSD4","ARF6","TTC8","TMEM63C","NOXRED1","AMER2","AKR1E2","KBTBD6","OTX2","FAAH2","NUDT5","BEND7","ATP5F1C","PRPF18","TAF3","VSTM4","VDAC2","COMTD1","SLC18A2","PDZD8","ZNF503","ABRAXAS2","QSOX2","FAM204A","NSD1","PRDX3","ENOX2","GHITM","CLEC1B","SNAPC4","TMEM52B","PMPCA","ENTR1","AK8","SPACA9","TSC1","GFI1B","HPRT1","BORCS5","DIPK1B","ZMYND19","STOX1","RET","DDX21","BMS1","STK32C","JCAD","OR4K2","FUNDC2","PIP4P1","METTL17","NDRG2","RNASE7","ARHGEF40","NSMF","ZNF219","C12orf50","CASP7","PPP1R36","BTNL9","CCDC186","VWA2","METTL3","SALL2","TRUB1","CYP2C19","ZFYVE1","C10orf82","HSPA12A","FRAT1","UBTD1","E2F7","ARHGAP42","ISCA2","LARGE2","PACSIN3","TTC7B","SLC39A13","PSMC3","RAPSN","AGBL2","TC2N","CPSF2","MOAP1","IFI27L1","IFI27","CLMN","CCDC38","PTER","C1QL3","CACNB2","HACD1","ARL5B","SMCO4","CEP295","TAF1D","ABTB2","R3HCC1L","AMOTL1","HTRA1","LIPC","CEP57","TCP11L2","SPRED1","TMCO5A","JAM3","GLB1L3","SVOP","GPT2","AMN","RAB8B","IKBIP","RPUSD2","HIF1AN","NDUFB8","ZFYVE19","SPINT1","FBN1","DEPDC4","BRD7","CKB","TRMT61A","BTRC","POLL","BAG5","DPCD","LARP6","API5","ASPG","ZNF319","HPS6","SENP8","NOLC1","ALKBH3","COPS2","GABRB3","SGPL1","FRS2","CCT2","PCBD1","ARIH1","C16orf71","CLMP","SCN3B","COX11","ZNF202","FAM227B","STXBP4","CYYR1","CUL5","MYRFL","WBP1L","BORCS7","C2","PLEKHF1","ANAPC16","SMPD1","APBB1","SYNPO2L","NUDT13","C11orf65","TRIM44","ILK","TAF10","TPP1","DCHS1","MSS51","CYB5A","USP54","RAG1","C11orf74","WDR88","ATP9B","PPFIBP2","CYB5R2","SERPINB7","KIAA0355","SERPINB8","TUB","RIC3","IDH3A","WDR72","PLD4","ZMAT1","XRRA1","TRIM66","RNF169","RPL27A","DENND2B","CDYL2","TMEM130","CENPN","AKIP1","ATMIN","C16orf46","TMEM41B","LEO1","ZNF143","TMX3","WEE1","MAPK7","PRKCB","HDGFL3","NDST2","MCM7","CLEC3A","CCDC68","CLEC4E","ZNF3","CLEC4D","ZSCAN21","RIMKLB","A2ML1","BEAN1","TK2","TMED3","SEC11C","CPLX4","GALR1","TMEM135","IQCD","NDEL1","CENPV","CDH16","RRAD","CIAO2B","CFAP52","HSP90B1","BLCAP","CHRFAM7A","ATF7IP2","MMP10","TVP23A","BEX3","TMPRSS5","COG1","PLEKHA7","ZNF606","B2M","ZNF592","CASC4","NNMT","ACSM1","AP1G1","SLFN5","CATSPER2","BMERB1","MARF1","SAAL1","PPIB","LDHC","CIAO2A","LDHAL6A","FAM111A","PCLAF","KIF7","LDHD","PLIN1","PEX11A","TMEM170A","MESP1","ANPEP","SCNN1G","NAV2","ANKDD1A","GLYATL1","C18orf54","DCTN5","TERF2IP","PLK1","CLPX","ZBTB39","CACNG2","MYO1A","CHP2","NEMP1","NAB2","VPS39","STAT6","PATL1","ATP23","STX3","MRPL16","PIP4K2C","MTMR10","YWHAB","C15orf48","GREM1","NYAP1","TSC22D4","MS4A7","MS4A14","DIS3L","CCNDBP1","EPB42","SMAD3","MS4A8","CCDC178","MAP1A","RCCD1","AKTIP","MAPRE2","EVA1C","MARS1","MBD6","CNPY4","PDIA3","NUDT21","NAT16","TERB2","NKX3-1","SGSM1","DUSP18","TEF","MEI1","B4GALNT2","PBX3","GNGT2","PHB","SNRPD1","TTC16","SAMD14","PIP5KL1","TMEM92","FAM102A","ACSF2","GOLGA2","TRUB2","COQ4","SLC27A4","ANKRD40CL","URM1","CERCAM","DOLPP1","CCDC103","ENDOG","PRRX2","UGT1A6","C15orf39","ISLR2","SP2","PRR15L","COQ7","GPRC5B","CRK","FBXO22","TBC1D2B","NOD2","SNX20","LOXHD1","KATNAL2","HDHD2","ZNF91","IGF2","RNF214","CDK12","DPEP2","DUS2","POP5","ENGASE","RBFOX3","ATP5MG","CD3D","TBC1D16","TEPSIN","MYO5B","ACAA2","STIM1","RRM1","OR51E2","TRIM68","OR51B5","FN3K","PRRT2","ZNF23","IRGQ","ZNF226","ZNF180","PPP2R3B","ZNF668","ZNF646","VKORC1","LPO","CA4","SMG8","TPM4","RAB8A","GPX4","MIDN","JSRP1","NIBAN3","KLHL26","GATAD2A","MVD","CDT1","TRAPPC2L","ANKRD11","SPATA33","RSKR","PROCA1","RPL13","ZNF641","CACNB3","DHRS13","TP53I13","KMT2D","CORO6","RHEBL1","TUBA1A","TUBA1C","ZNF610","ZNF528","ZNF701","SERTAD3","NCKAP5L","RAB4B","AQP2","GPD1","PROSER3","CYP2S1","AXL","NFKBID","TMC4","LAIR1","TTYH1","LENG8","CDC42EP5","TMEM145","ZNF526","TRAPPC9","ZNF146","ZNF283","SPINT2","C19orf33","YIF1B","DNAAF3","PSCA","ATCAY","LY6D","DAPK3","EEF2","TMIGD2","CHAF1A","UBXN6","HDGFL2","PLIN4","SEMA6B","ZNF444","NXN","TLCD3A","GLOD4","MFSD3","GPT","KIFC2","SLC43A2","RILP","SERPINF2","WDR81","SRR","TSR1","TRPV3","HSD11B1L","CYB5D2","GGT6","C19orf48","KLK4","KLK2","KLK6","KLK11","KLK13","ZNF83","KRT80","OTUB1","RCOR2","ANGPTL4","CD320","SPRYD3","ZNF558","NDUFV1","CDK2AP2","NUDT8",null,"PRDX2","ZNF232","MIS12","CD300A","TEKT1","HID1","MRPL58","ATP5PD","TMEM88","EVPL","SRP68","TMC8","TK1","TMEM68","GSDMA","KRT24","GHDC","FAM234A","ZNF598","RAB26","MLST8","E4F1","DNASE1L2","ECI1","CASKIN1","ABCA3","KCTD5","SRRM2","ZNF597","NLRC3","SDHAF2","DDB1","VPS37C","VWCE","RAB3IL1","BEST1","FTH1","BSCL2","POLR2G","SLC3A2","SPINDOC","ATG16L2","C2CD3","TRANK1","TTC21A","RPSA","ENTPD3","CTNNB1","ULK4","FADD","LTBP3","SAC3D1","BATF2","SLC22A11","SF1","MAP4K2","CCDC88B","SCARA3","PNOC","COPS6","PAFAH1B2","ANKS3","NUDT16L1","KIAA1586","RAB4A","SETD5","VASN","FAM83B","THAP9","RNF187","HOOK3","MAPK1IP1L","DDIT4","RBPJ","LMBRD1","ZCCHC4","PTGDR","TTC39C","GLYCTK","GNG4","UBTD2","POLR2J3","NKIRAS2","DNAJC7","IRF2BP2","NT5DC2","FOXI1","SMIM4","COA6","KIF5C","MGAT2","BMI1","THAP11","MMADHC","PDHB","PXK","H1-4","PCMTD1","KCTD6","MPLKIP","ACOX2","FAM107A","IRF2","MOBP","CX3CR1","XIRP1","DEGS2","SCN11A","ARF4","SEPTIN2","FILIP1L","MFSD2A","DTYMK","TAP1","ING5","ATG4B","BDKRB2","MLKL","RFWD3","MTNR1A","KCNG4","RHOH","COG7","CDC40","STIP1","SCNN1B","HR","TXNDC2","RAB31","REEP4","TNXB","BMP1","ATXN2L","CCDC110","POLR3D","FEN1","CAVIN2","MTCL1","HEXIM2","FNTA","SERINC2","TRAPPC11","ING2","CDKN2AIP","SNRNP48","TMEM223","SLC20A2","DYNLRB2","TMUB2","ADAM29","STAT3","ZSWIM1","ADAM9","GDNF","AXIN2","NDUFS5","VWA3B","ZNF30","LRATD2","LDLRAD4","KCTD19","SLC16A4","IL7R","TMEM208","LRP1B","WFDC12","AHCYL1","DNAJC21","PKIG","NPNT","FAM178B","SEMA4C","CNNM3","GSTM4","TET2","CXXC4","TCTN2","PPIP5K1","TSPAN5","ABHD15","ZBTB5","CHTF8","ADAL","LCMT2","SNTB2","IL12A","ZNF507","STX18","NSG1","ZBTB49","GFM1","DDX19A","ATOH8","ANKRD49","SFTPB","USP39","TNIP2","C2orf68","TMEM150A","RNF181","VAMP5","LRRC28","MAT2A","PLA2G4F","ENHO","ZNF608","SLC35G2","INPP5D","LETM1","TMEM129","PPIC","SPRY3","CEP120","STXBP6","TM4SF20","MFF","GRM5","LGALS9","JMJD7-PLA2G4B","PXDC1","SIGLEC7","E2F6","FEM1B","COMMD8","ATP5ME","UQCRFS1","SLC49A3","COL4A3","MAP2K1","KLK7","HNRNPH1","IRS1","MECP2","UPF3A","ZBBX","AR","DHRSX","VXN","HSPBAP1","ASMTL","SLC25A6","CHST14","PARM1","CSNK1G1","FAM110B","ARMC4","AFAP1L2","ZNF354A","ATF5","UBE2V2","ZBTB43","CPT1C","PCSK9","XPO6","GSG1L","MN1","APEX2","NSMCE1","CCDC126","NPIPB12","OR10G3","RAB3B","CD2BP2","RGS14","TBC1D10B","LMAN2","GCSAML","RAB24","PRELID1","THBS3","CA5B","SLC50A1","EFNA1","CXCL10","NPIPB3","SH3TC2","CXCL11","ZRSR2","NMD3","ADRB2","B3GALNT1","GPRIN1","KCNAB1","MRPL1","SHE","PGM2","STK32A","C22orf15","OR5AU1","MINAR1","GP2","SLC33A1","SNUPN","CRADD","SIN3A","ARL13B","PTK2","RSPH10B2","PTAFR","PTPN9","RNASE6","CXCL8","SCN9A","RASSF6","SDC2","CD52","MMGT1","SPRR1B","SPRR1A","TM2D2","HTRA4","PLEKHA2","CLIC4","SLC38A11","GPR183","CRCT1","CCDC8","METTL15","MUC15","ZEB2","GJB1","PCBP1","HINT1","DTWD2","CLIC3","INO80E","BNC1","DFFB","NFU1","ANTXR1","CKAP2L","C15orf40","RAMAC","APLF","BOLA2B","RGPD8","HIC2","LUZP1","HEXD","BUB1","SPNS1","LRRC45","CHRNA5","CENPX","AGPAT2","ASPSCR1","FASN","CNBP","MT1E","DUS1L","GPS1","RFNG","DCXR","ZNF32","LDB2","RAC3","NRG4","LIMS1","TMEM266","TAPT1","UGP2","HNRNPF","BTD","CSGALNACT2","PCDH7","ROBO1","ONECUT1","AVEN","P2RY1","TRIM56","MUC17","REPS2","MUC3A","SYAP1","ITGAM","PYDC1","TPST1","TOR1AIP2","TM4SF1","OTUD3","OTUD7A","GUSB","BRD3","KLF13","ZFPM2","ZNF764","ZNF747","ZNF768","TAS1R3","TMEM42","MAP3K2","PUSL1","SF3B5","ZNF35","TIGD4","IFFO2","NLGN2","MYO7B","CHD3","TMEM154","MYRIP","ALCAM","YWHAG","UBE2E3","CNTROB","TRAPPC1","KCNAB3","FAM153A","GPR37L1","SIMC1","TMEM192","SPDYE5","ZNF778","NIPA1","GPR25","UBE2E1","HNRNPA3","SIK2","RNF150","CCDC144A","CHRNB1","GYPA","USP38","SLC16A5","NANP","ANKK1","ADPRM","FABP6","PWWP2A","USP50","USP47","PDCD6IP","ZNF212","FAM161A","ZNF282","GLB1","GON7","FAXDC2","CRTAP","CNGB3","ELP5","CMTM8","GABARAP","LGALS9B","STX8","CDK1","UBB","NFRKB","FRMPD2","PRDM10","B3GNT2","FOS","TMED10","SETMAR","SMAD1","EMX2","TCAF2","SEMA3E","LRRN2","SLC30A1","DCLK2","GPRC5C","TMEM182","VSTM2A","KRT8","KRT78","ADORA2B","MGMT","KRT86","HARS1","NFXL1","DENND5B","CD14","DNAJC18","RIOX1","SPATA24","RALGAPB","PYM1","MZB1","KRT4","SLC23A1","NPAS2","LONRF2","NUDT9","HSD17B13","PA2G4","ELOVL6","PFKFB3","TMC7","ARL6IP1","SERPINB9","SMAGP","EMB","SIX2","DLGAP1","STAT2","NUDCD2","IRF2BP1","HSPA4","FOXA3","SLC26A5","COMMD5","GTSF1","ZNF16","ARMC10","RNF34","ACYP2","TRABD","ATF7","RASA4B","SOCS6","ZNF296","TTLL6","POLH","KCNS3","KIF5B","AKAP13","CDCA4","SDR16C5","DYDC1","OR10A2","CHCHD7","FOXN2","LMOD2","USP32","CEL","PPM1D","GPR27","LOC93622","KBTBD2","RIOX2","TRIAP1","LSM3","KIAA0232","MTSS1","TMEM43","RNF139","RPS9","TSEN34","GSTA4","MSANTD4","NDUFA3","OSCAR","PAQR8","NUDT6","TANC2","OR7G2","PKHD1","OR1M1","DNAJC24","MBD3L1","ZNF160","ZNF415","CAVIN3","CEACAM3","DCDC1","HAS2","PDGFD","S1PR1","HS6ST2","PYGO1","LRRC8E","PKIA","XKR6","TSNARE1","FPR2","FPR1","FEZ2","SOX7","C8orf74","C11orf24","ALK","KYAT1","MTM1","TRMT61B","INSR","MFN1","GIMAP8","NRTN","KCNMB3","FUT3","KCNG3","ATP6V0E2","PRKCE","JAGN1","TADA3","SOCS5","C1GALT1C1","C9orf16","MORN4","ZNF672","ZNF692","NAIF1","RBKS","OR2M4","GRIK1","MUC7","TMEM126A","TMEM126B","TRIM8","NETO2","CLDN20","CDC42BPG","SCAND1","JUNB","FAM241B","TMEM37","UGT2B7","LRG1","SHCBP1","SOSTDC1","NPTX1","FAM98B","ZNF439","ZNF440","GAA","CANT1","KCNK3","ZDHHC16","CHST11","EXOSC1","PGAM1","CHD7","ESCO2","KRT19","KRT15","LURAP1","CLCN5","TPPP","KCND3","KRT13","PDE7B","MRPL36","ZNF581","NAT1","KRT20","KSR2","ZNF524","MCC","ZBTB26","DSEL","POLR1C","ASXL1","DLK2","ZNF562","ZNF318","ZNF561","WIPF2","HOPX","LRRC8C","RSL1D1","LRRC8D","PPID","COL24A1","ETFDH","LPAR3","PTGER4","TBCA","NEUROD2","MAP6","BCL2L1","OR2AT4","PLRG1","ZNF584","DNAI2","NMUR1","CLSTN1","CXXC5","ZNF274","PIK3CD","SLC25A33","ENC1","SPSB1","P2RY6","BPTF","ZIK1","GPR82","GPR34","PLEKHG5","ATF7IP","LKAAEAR1","RGS19","TCEA2","DEFB4A","HDAC3","SPATA46","GPHN","TMEM51","CAMTA1","LGALS4","LRRC34","SPATA5L1","GATM","RASGRP4","NHLH1","SLFNL1","BCL2","RHNO1","CTPS1","KNDC1","METTL18","CFAP46","COL8A2","PWWP2B","PCDHB1","ZNF540","ANGPTL7","FBXL14","EXOSC10","ZNF570","NINJ2","MLLT3","FAM90A1","RRM2","TRAPPC12","RPS21","C3AR1","MRM3","PTEN","RPS7","PRND","RNASEH1","PRNP","KLF17","FRMD5","CYP4F11","TLN2","LGALS9C","TVP23B","FBXW10","ZNF217","SRGAP2C","ATPAF2","FOXB1","PPIH","DRC3","ZNF57","SHLD1","JMJD1C","LDHAL6B","SYNPO","ZNF556","MAL","ZNF554","RAB33B","THOP1","ANKRD20A4","LAMB2","USP19","QARS1","ORMDL3","SERF1A","KLF11","SMN1","EIF2AK3","MOB3A","KRCC1","NME6","CYCS","CD8B","SLFN12","SLC9C1","OR1A1","FRMD3","SNTB1","MTBP","TEFM","MRPL13","MALT1","ISG20","MBOAT1","ID4","GPR22","CXCR6","CEBPB","AZU1","TPSAB1","PAIP1","CLEC7A","C5orf34","NEGR1","ZNF131","MACROD2","DPAGT1","BSG","HINFP","CERS6","SPTLC3","COPRS","TP53RK","B3GALT1","OR5A1","CLEC12A","OR5A2","BPGM","POP7","ALG14","SUCLG2","STARD5","CSDC2","RCAN2","IL16","GNB2","CFAP53","OR5B2","MCRIP2","PDZD3","C2CD2L","ARNT2","GNG12","PRSS27","SYNPO2","DNAJB7","CLP1","EFCAB3","TTC36","RSPH9","COPS9","GTPBP2","FGGY","IL17D","FUT9","TCEAL1","ZNF24","HSFY1","MANEA","RAB40A","MAB21L4","AGXT","AFF1","FIBP","CARNS1","OR10H5","BANP","PPP1CA","HCFC1","CTSW","NIPAL4","MUCL1","SNTG2","PDE3A","RASGRP1","KLHL6","CHCHD1","MRPL52","SMPDL3A","RND1","RAD9A","EFEMP2","AGAP5","WASHC2C","TMEM134","ZMAT3","ZFAND4","THEMIS","ZNF738","MS4A10","SLFN11","FAM71D","CORO1B","FUT10","LRRC20","MUS81","TMEM217","OR4D9","ZNF596","CFL1","TMCC1","NAA16","EFCAB12","PSME3IP1","RAB43","FADS6","CBWD1","RAB37","DCP2","SNX32","RPL38","CYP7B1","OVOL1","RARG","CES4A","CES3","SSH3","CES2","PDP2","SP3","DMXL1","METAP1D","ZNF621","EGFL7","NADSYN1","DHCR7","NBEA","RNASEH2C","MYEOV","ANKRD13D","MYD88","OXSR1","SLC22A13","PHF8","LCLAT1","ADH6","KAT5","SH3RF3","GXYLT2","DCAKD","TADA2B","CCDC96","GRK2","RELA","EVC2","ZNF680","HECTD4","FAM222B","BNC2","HPSE","COQ2","HSPA6","TRMT112","LRRN3","KDM2A","ACSM6","ADCK5","MRPL57","NOC3L","ESRRA","RHOD","COMMD1","RAPH1","MTX1","ADCY5","PARP14","CYSLTR1","PARP15","CKS1B","ABCD2","ABLIM3","MAB21L3","MFSD4B","VANGL1","GLRX","IQCB1","SYT12","GOLGB1","DMRT2","ZNF483","SLC2A14","GPR137","SNCG","MMRN2","MZT2A","TNKS","ZNF449","ZBTB21","PPP1R3B","OR10K1","STOX2","MAP3K11","TRIB1","KCNK7",null,"C1QB","C1QA","NDNF","IQCF1","OLR1","GLIPR1L1","DAG1","DAB1","ARV1","NAA20","CCDC36","SAA1","MICOS10","EHBP1L1","THAP2","RNF26","PPP1R14B","ZNRD2","AGR3","SMARCC1","ZNF417","PTPRM","FKBP2","VEGFB","PEAK1","TNFRSF10D","MST1","TNFRSF10C","GMPPB","MOB1B","ZNF622","CSPG4","SNX33","FAM166C","NABP1","ADGRF3","CHD2","XCR1","CCDC106","CCR9","CEP83","SULT1B1","NUDT4","PC","UGT2A1","SCAI","NMNAT1","LRFN4","TRAPPC3L","APOBEC4","SLC19A1","HSPB7","RCE1","UQCRH","TAS1R1","EIF1AX","SPDYE2B","BCLAF3","PSMD1","ADGRG2","MUC13","SUSD5","HEG1","C11orf80","TOMM20","C1orf100","AGFG1","STAT5B","CD7","CNP","JUP","HAP1","TDRD12","EIF1","ENDOV","RNF213","TIGD3","KCNH6","MARCHF10","PLK3","NET1","DPY19L1","DET1","PHOSPHO1","ZNF791","PHC3","GPR160","SPTBN2","GOLIM4","RBM4B","ATP5MD","HOXB2","C1QTNF1","MARCHF3","SWSAP1","SLCO4C1","RBM4","PIFO","XXYLT1","UBXN2A","RAX2","LRRC63","CCS","NRROS","CEP19","KLHL15","FBXO45","GNG5","SLC25A30","C9orf131","CD34","CTSF","MSRB3","LEMD3","C16orf91","TLR10","TLR1","TLR6","FAM174A","RGMB","FAM53A","CYB561D1","GSTA3","ZDHHC24","TRMT10C","SELP","CTU2","MGA","C12orf66","ARL13A","SNX31","PIGG","PRPF8","ADCY6","PITPNA","DDX23","ZNF80","ZNHIT2","ZBTB4","TNK1","ZHX3","PHLDA3","DIRC1","SLC16A11","SLC16A13","CHRNA9","C11orf45","EXO1","RALGAPA1","LIG4","GTF2IRD2B","ABRA","ATP2A2","ZWILCH","RPL4","SNAPC5","C12orf76","CNTNAP2","BBS1","DENND4A","GCSAM","ANKRD36C","SLC26A9","MFSD4A","PELI3","MYO1H","MRPL11","IL20RB","GOLT1A","AKIRIN1","MSL2","ZNF497","CMKLR1","ANGEL2","UGT8","KY","IQCK","SLCO2A1","ZNF266","OR7D4","SLC29A2","BRSK2","B4GAT1","TMEM167A","SH3PXD2B","RESF1","LARP7","FGFBP3","NR1D2","BRMS1","RPL15","FAM241A","HRAS","WDR49","SRP72","RIN1","ODAPH","THAP6","CEP135","FZD4","BTC","PDZK1","ADGRE1","DENND6A","PDE12","GLMN","DNAH12","YIF1A","CNIH2","AMY1B","NLRP6","NDUFA11","RSRC1","CATSPERD","RAB1B","PTDSS2","MICOS13","C3orf33","SEZ6L2","KCTD13","P2RY14","AMZ1","GPR171","CD164L2","FUT1","DHX36","FBXW8","ZG16","KLC2","SLC22A1","CTBP2","CHST2","ZDHHC14","ATR","UBE2C","GK5","VCPIP1","PDIK1L","TRAF6","ZNF654","TVP23C","MRPS22","PACS1","MARCKSL1","SH3BP5L","YPEL2","CADM2","ABO","PSMD2","PPM1E","FAM131A","CSRP2","PARL","DDIT3","PCCA","DCTN2","ZNF408","CTDSP2","CKAP5","ARHGAP1","MED16","ATG13","C1orf127","CHST1","GOLGA8A","VWA3A","TP53I11","CENPS","DOLK","PHYHD1","CCNE2","PHYKPL","ANKS4B","CST6","GRAMD2A","ZNF519","LSM1","BANF1","APOF","CHRNA7","TMEM9B","NRIP3","PTPN2","SCUBE2","EIF1AD","SMAD2","EIF3F","ZNF25","ARL10","CLTB","PCSK1","RFESD","CCDC14","TBC1D10C","SART1","PPP2R2D","MCTP1","POLD4","LRRC25","DPP10","CLCF1","UBQLNL","UBQLN3","LIPT2","KCNE3","CABP4","ALG10B","DRAP1","LONRF3","UCP3","UCP2","C11orf68","PAAF1","MRPL48","RAB6A","P2RY2","FOSL1","ERCC4","CCDC85B","TMEM70","RPS6KB2","RMI2","TOM1L2","TEX26","ZNF77","GPR156","MTLN","KDF1","B3GNTL1","MLXIP","NR2F1","AURKAIP1","TTLL11","TOMM5","SLC35E3","ZNF169","RUVBL1","SFN","MSRA","CTDNEP1","ETV4","SWI5","GAPT","BAIAP2","CREG2","ZDHHC21","PLEKHF2","A2M","ARL4D","DOK7","LRRN1","UBE2O","ORAI3","UNC119B","DENND2C","PLEKHD1","ASCL3","TUBB6","LYSMD3","AMIGO3","B3GALT6","ZNF613","C11orf16","TMPRSS7","NUPR1","JAKMIP2","MBLAC2","TPRN","ZNF683","SLC35A4","CRYBG2","IP6K1","SSNA1","CSTF3","YES1","CHMP6","TMEM39A","TCP11L1","GPX2","CCDC57","FOXG1","SPHK1","BNIP3","MYPOP","LRRTM4","ATAD5","SMIM19","ZNF404","RTTN","OR4K17","ACBD7","ANAPC2","OR4K13","ZBTB8OS","SLC35G1","ZNF135","COX8A","TAC4","ZSCAN2","PRR18","B3GNT4","CDC26","HSD11B2","CRLF3","RNPEP","EID2","DMRTA1","EID2B","RIMS2","KCMF1","DNAJC30","SPRYD4","SYNE3","CLK2","LPCAT4","SLCO3A1","ZNF575","WDR25","SGF29","PLAAT3","OR5AN1","PHLDB3","PRR15","GNG7","USF3","CNTD1","KBTBD11","B3GNT5","MAP3K19","LMNB2","RMDN1","MEX3C","RNF152","MYO1D","LRRC37A","OR4F17","BDNF","CCDC121","ACSF3","BOK","ZNF843","RBIS","PFN4","CDK5R1","NCKAP5","RUFY1","BASP1","OR51L1","LRRC37A3","VSIG10","IRX5","METRNL","FAM91A1","WSB2","GRIN1","SOX11","TYMS","PXMP2","TCEANC","PNMA1","TCIM","MAMSTR","ANKLE2","FUT2","EFCAB5","GCNT4","MUC20","THAP4","NFATC2IP","LY6H","FIBIN","FAM89B","SHMT1","DPP7","SEC24C","SMCR8","MTHFR","C19orf18","DEAF1","MTX3","TMEM80","SIX5","IFNW1","FBXO46","ZDHHC13","SLC38A9","ACER2","WDR73","POLE","PHETA2","SCN4B","DSCAML1","RHOG","EPS8L2","ZDHHC22","ANO6","ZBTB34","FAM9B","NUDT4B","FAM210A","TALDO1","ULK1","RIMKLA","CLVS1","RPS6KA3","B3GNT8","PUS1","CHD9","OR2T12","SPACA4","OR2T33","GATD1","TRIM72","MAN1B1","KCNA3","FZD8","GJD4","FBXO39","KCNA2","TOP3A","CASKIN2","ZBTB38","BEND2","CCDC71","LRRN4CL","TIMM22","HIC1","PPFIA3","MAGEF1","UMODL1","SAMD9L","PAWR","TGIF1","MIEF2","NAP1L5","NIM1K","CD19","ERICH5","NR2C2","GPR4","ACOT4","CAVIN1","ARIH2","RBM44","ZBTB33","ZBED2","IRX3","RPRM","OR2B11","SLC25A22","RABEP2","ATOX1","TBL1XR1","SAMD12","CD163","C18orf32","PIDD1","ZNF491","RPLP2","HASPIN","JUN","CSTF2T","PGBD5","C12orf54","GBA","ACAD9","IL17RA","PNPLA2","AGTRAP","CD163L1","SRRM3","THAP5","CRACR2B","SUMO4","MAGEB10","DNAJC28","NAALADL2","CD151","POLR2L","FAM20C","NECTIN3","SLC35G5","ANXA2R","TMEM94","FLII","SOX12","HNRNPA0","KCNJ10","CHID1","PCDHB9","ZNF620","ZNF518A","TMEM187","SVBP","ZNF619","CCDC184","AP3S1","GRB2","ZBTB41","UBE2N","ARL6IP6","ZNF354C","MAMDC4","CENPBD1","ODF3","BET1L","RPS27","RIC8A","IMP3","ASB8","LCN15","ODF3B","DPY19L2","C2orf73","TSPYL6","LRRC75B","DMAP1","ADAMTSL1","CALHM5","IMPDH2","ALS2CL","MLF1","NDUFAF3","C2orf69","GRAMD1C","STAP2","TSSK6","BOLA1","PDE4DIP","DDX10","GOLGA8Q","PPP1R42","NDUFV2","DALRD3","ZNF114","ZNF518B","AMER3","ZNF366","LCORL","PARD6G","ZNF454","SH2B1","ZC3H12D","VN1R1","POGLUT3","PLEC","RNF212","PRSS36","ZNF543","TMEM151B","GALNT11","WDR6","GEN1","TMPRSS9","AQP11","TMEM11","ZNF354B","KCNG2","CALML3","ZFAND2A","PLEKHM3","ZNF223","FAM220A","DNAJC22","CEP295NL","BEND3","NT5DC1","GLDC","COX14","MCMDC2","P4HTM","UCN3","DTX3","KLHL11","CTXN1","SLC25A20","CA8","CD28","EPM2AIP1","ERBB4","MAF","CTNNBIP1","GTPBP6","ERN1","GPR35","C10orf53","CSRNP3","ZNF713","PARP10","DYNAP","SUZ12","NSUN3","KCTD12","DHFR2","RPP25","GRINA","THBD","GP5","COX5A","STX19","FAM219B","ZHX2","CPN2","CPNE7","CD300LB","GDPD4","RIIAD1","MPI","TRIM73","OPLAH","TMEM52","TMEM139","EFCAB13","MSC","APOLD1","RFLNA","EXOSC4","DPY19L3","TAF7","ZNF852","FOXE1","PFAS","HYI","CYBC1","TPRX1","LGALS7","ZNF552","GAK","ZBTB7A","TUFM","ERICH3","RMI1","CTC1","FBXO34","SELENOW","EIF3K","MRFAP1L1","SNX18","AURKB","C14orf39","MRFAP1","C3orf38","KLHDC7A","TMEM107","RRS1","EXOC3L1","TRIML2","RCC2","IGSF22","C9orf50","ZFP42","CCDC89","DPM3","CYC1","PER1","TMTC2","FARSA","SPTY2D1","C10orf67","SAMD4B","GIMAP7","EDC3","TCAIM","FUCA1","GGN","TMEM125","ZNF664","CALR","MAGED1","GVQW3","LDLRAD3","CDH4","RAD23A","PCARE","GADD45GIP1","DAND5","PTPN11","NSUN7","FAM156B","RAB39A","CLK3","HLA-DQB1","GATA2","ARID3B","PACS2","ELMOD2","EGR3","CATSPERE","VWA1","GEMIN4","KLHL28","MKRN3","ZBTB18","C14orf28","SHARPIN","LBX2","DNHD1","SLITRK4","GCC1","LSMEM2","CIITA","ZFPM1","ALOX15B","PLD6","CDC42EP4","OR6C4","ZBTB42","LACC1","MAF1","FCER1A","WDR97","PCED1B","APOBEC3B","PIPOX","FAM216B","MYADM","MROH1","SERTAD2","AKAP5","NKPD1","RNF227","CITED4","ABCA13","TIGD5","PDXDC1","C1orf194","ZNF154","R3HDM2","B3GNT3","NRXN1","SEPHS2","GPBAR1","ZNF784","ZNF648","C14orf119","GOLGA8J","BBS10","FIZ1","PUF60","SSC5D","DCTPP1","ZNF771","TSHZ1","PSTK","SOCS4","ZADH2","ZNF48","ANKRD18A","WFDC11","TMEM86B","SEPTIN1","TRNAU1AP","EXOC3","TDRD6","C12orf40","CSNK1A1L","LYNX1","MED14","FAHD1","TDRP","RCC1","PRKRA","ZNRF2","RRH","SLC9A4","ZNF816","FGD6","ADGRD2","PLD5","OAZ2","PNPLA1","CCDC43","KCTD4","MEIOC","FZD2","TIGD2","ITPRID1","HCLS1","MTURN","ZNF609","PAK2","CCDC66","DEFB124","MCFD2","HARBI1","C11orf71","CYP8B1","GAS1","ARHGAP45","ZNF571","GLIPR1L2","MIGA1","KCNE1","PRR26","NRIP1","ZSCAN4","RNF182","TSPYL5","FUT7","H2AC6","SKIDA1","H2BC4","MB21D2","SSTR2","ZNF594","PCGF5","SLC47A2","PRF1","OR2A4","MAB21L1","YOD1","TMEM64","SHISA2","S1PR5","GPR157","SLC36A4","ZDHHC20","ANKRD30B","ZFP3","ARSJ","MAP3K15","PPA1","PSMG4","MAP6D1","ZNF443","CXCR2","GREM2","SSR4","CAPS2","ZNF792","CUEDC1","SCRIB","KCTD2","D2HGDH","OXTR","CMTR2","FAM83H","OR56A1","ZNF572","ST20","PITPNB","TCEAL8","OR52E4","LRRC57","MRPL14","GPR137C","BBS12","ZFP82","OR52N5","LSMEM1","NQO1","AEN","FKRP","TRAPPC5","RPH3AL","SLC25A42","FCRL6","METTL23","SLC26A11","HIGD1A","MAPK15","EHMT1","ADIPOQ","F2R","ZNF707","MUC16","NPM1","PJA1","DHTKD1","C4orf50","H2AW","ZNF746","POLR2A","TLCD5","FRAT2","TMEM102","ZNF322","NME9","HEPHL1","LRRC75A","CFAP65","DDX60L","SYNE4","OGFOD3","WASHC1","AATK","DDN","ZNF467","SOX2","ZNF678","TMEM45A","RAP2B","ZBTB2","RNF135","ACBD4","SGSH","MAB21L2","FANCB","SETD2","C6orf223","MEX3D","MRPS23","FDCSP","GPR135","SLX1B","ANKRD62","P2RY13","TNFSF15","ZFP41","PHLDA2","ATG9B","ZNF875","PLAG1","OR8H1","YIPF6","OR5T2","ZBTB20","DIPK2A","C5orf30","AMIGO1","GPR3","OR5AS1","SIAH2","COPG1","SLC9A9","LSM10","RELL1","RFX7","SLC35C1","TIGIT","RNF41","SLC2A4","IBA57","CLDN7","ZNF329","ZNF101","C5orf24","ADO","COA4","OR4P4","PRKAG1","GINS3","CCDC149","MRPS11","SNRPE","RTKN2","PNMA8A","CHST15","USH1G","MGAT4C","IDH2","OR52A1","TMEM259","GET1","TNRC18","FAM181B","TMEM30B","DEXI","ZNF716","NOP10","TDRKH","ZNF708","IST1","ERCC6L2","MRPL41","CREB3L2","P2RY8","UNC5C","TSEN54","RGMA","ASB18","UBA7","MRPS16","SHISAL2A","RAD51B","LDOC1","ARL6IP4","EXT1","SHMT2","MOB2","HHIPL1","ATP6AP2","CYB5D1","FAM153B","BACE2","UBE2E2","SYNM","GABRG3","NLRP10","IZUMO1","AP1S2","C8orf33","DCAF4L1","SPACA6","ZSCAN22","KCNJ14","FBXL6","C1S","GLTPD2","KIAA2012","KBTBD3","YBEY","CLN8","PLCXD1","CACNB4","TRAPPC6B","PGBD4","NPLOC4","TSHZ2","CAPN12","EXOC7","KPNA2","XKRX","CEP97","FES","GLRX5","FAM104B","MXRA7","LIMK2","MFSD5","RNASE10","ADI1","RWDD4","SPNS3","SATB1","NXPH3","CSF1R","EPHB3","ACTL10","EPGN","SNORC","TRAK1","TSPAN10","OR2V2","PLCB1","SKA2","NDN","TTC3","BRICD5","IGIP","TSKU","CMC4","ANXA2","PAQR7","PAPPA","MAFA","NGRN","RPS17","HCAR2","CCDC87","C1orf116","CRIP2","DDX28","ACBD3","C16orf72","PLCXD3","VMO1","OR4F15","ALG12","LCK","COL18A1","RBM10","ADGRG3","RPL35A","SLC25A18","ZNF721","C11orf54","CEP63","SRPRA","EWSR1","HMGN4","SPATA13","GJC1","CNOT10","MTA1","ZNF662","CADM1","ZNF320","C12orf60","PYCR1","NAA38","SPNS2","MCEMP1","AP2A2","SLC8A1","SLC25A21","ABAT","SLC25A10","CAMK1D","RGPD6","LYSMD4","WBP2NL","IGSF5","AFMID","GAS6","NEB","BEGAIN","ARHGEF37","CSMD1","CEP57L1","RIPPLY3","ANKRD20A2","GJD3","RABIF","TMEM119","FANCF","SMDT1","GABRR3","CHST6","POTEC","RUVBL2","GDPGP1","RIMBP3C","PRR36","PTTG1IP","DDX41","ABHD16B","CCDC60","PLGLB1","DAZAP2","CCBE1","SELENOF","TMEM121B","ZNF623","EPHA10","CCDC125","REC114","BOLA2","BCOR","JRKL","CABCOCO1","GBP6","KIAA2026","OVCH2","FHL3","OR56A4","C19orf71","CCDC159","RIPK4","NPIPA1","SF3A3","TRIM61","GRIN2A","GTF2H2C","ASB7","GPR132","MX2","EP400","TENT5C","COA5","UTP11","PSMG1","PRR14L","KLRC4","SERHL2","PCBP3","SETD3","TNFAIP8L3","ZNRF3","FBXL7","TANGO2","H3C13","SFXN4","MRPL54","ZNF438","HMCES","CCR3","DGCR6","GOLGA8G","RP1L1","C11orf88","ZNF530","NDUFB1","KLHL25","TRMT12","PSG9","BMP8A","ALYREF","RFLNB","EFHC2","UPP1","TRIM52","LHFPL6","CMTM4","TMEM50A","ASCL2","TBK1","CBX6","MACC1","ACSM2A","TBL3","ACP7","KREMEN1","TRAIP","CHEK2","AIFM3","B3GALT5","ZNF703","TUBA8","ELOA3D","NPIPA5","OLFML1","RBM12B","CCR4","LIN9","BTBD9","NUDT14","ANKRD45","MAATS1","GPR39","FAM3B","ZNF730","IQGAP3","TOB2","SCN5A","UTY","SRARP",null,"TTC32","LRRC55","DNAH2","SH2D1A","SDR42E2","PRKX","KMT5A","SMTN","PP2D1","COA3","NPB","ST6GALNAC3","PTP4A2","ACTG1","TMPRSS2","DENND5A","OR2T10","DIABLO","VPS33B","TBX1","ADAP2","UQCR10","FAM120C","EIF3C","NIPSNAP1","CNTN2","LRTOMT","KCNQ3","NR2C2AP","CRELD2","OR1D2","SCFD2","UBE2F","GPR173","PPP1R2","TSPYL2","GOLGA6L4","PGP","SNRNP35","IRAK1","CMSS1","PCDH9","ACOT1","OAF","ALDH1A3","CDR1","H2AC20","KCNK12","H2AC21","POU6F1","TM2D3","TSSC4","TACSTD2","CLECL1","SIX6","PRKD1","CCSER1","ZDHHC23","MROH7","SRPK3","SLIT3","EFNA5","H1-5","SPATA32","PKP3","MAP7D2","CSF1","COLEC10","PLA2G6","MAML2","SS18L1","TOP1MT","COPB2","LRRC19","THAP7","KNTC1","CCR10","NCMAP","WDR27","TXNRD2","C1QTNF8","FOXO4","PTP4A3","TMEM255B","PROS1","NUTM1","HDDC3","ZFP1","DUSP8","SOCS3","SLITRK6","LPAR5","XPOT","STING1","PDE4B","SNN","KCNH7","NELL2","KRBA2","MED12","ZNF93","SEPTIN9","CDCA2","AMER1","ZBTB40","H2BC21","CLDN6","OR51M1","SEPTIN5","EIF4ENIF1","LRRC26","SERINC4","RNLS","APOBR","FAM110C","ATL3","NDUFA12","TCTE3","UBE2G2","SATL1","OSBP2","ZBTB7C","APOO","TMED9","TMEM186","SDR42E1","RBM33","ARMCX2","BTBD6","H1-10","RBM43","SUMO3","IMMP2L","CLCNKB","JAG2","FMNL1","NUTM2A","PTRHD1","LCN12","WT1","ZFP90","FAM227A","MUC6","NOC4L","USP18","NDUFA6","CHRM5","SORCS2","TMEM121","TMEM106A","SIVA1","DGAT1","ROBO2","AP3M1","F8","NT5C1B","CA13","UBOX5","MAFF","BRF1","SEMA4B","SPDYE16","CIB1","ANKS1B","NELFA","SLC24A3","EFCAB10","FLRT2","INTS5","RPS27L","MANEAL","ADSS1","ANO9","FAF1","FAM43A","NSMCE3","HSF1","C6orf120","PURA","H2BC13","INPP5J","MFSD6L","LRRC37B","DDX51","NOMO2","SIGIRR","NRBP2","IFITM2","TNFAIP2","ZNF445","PGBD2","TCEAL9","RAB11B","PRMT3","GP1BA","PRPF39","PPIL6","ZNF74","KIAA0825","UBALD2","CDNF","KLHL33","RBM11","GALNT17","ZBTB37","IL3RA","CCDC137","RGPD2","ARL15","C12orf56","CDK10","SOCS1","TCN2","GAS2L1","ATP6V0A2","PRKN","TEDC1","HGS","TNFAIP8L1","OR2V1","RAD51D","OR7A17","MAPK11","SP140L","MRPL30","TARS3","SMYD3","METTL7A","IFNLR1","SH3BGR","FAM174B","ZSWIM9","KPNA7","TMEM179B","GPRIN3","KRT6B","PARPBP","STAC3","ROR1","MUC1","FAAP100","IRF7","L3MBTL1","BRCC3","SV2B","FAM131C","LMNTD2","PRKG1","NR2F2","TLCD2","LSAMP","AHNAK2","OLFML2A","SP1","MRPL40","INKA1","PCGF3","LMLN","P4HB","PSMD13","PBX1","NDUFA4L2","SHC4","ZFP36L1","UBE2L3","BRWD1","PMEL","SYN3","POU3F1","SNAI3","ZBTB3","MORN5","C6orf201","MYBL1","MOSMO","DRG1","ANKFY1","YTHDF3","ZNF696","ADARB2","SRL","IFIT1","CXorf38","ADAMTSL5","KCNIP4","MORF4L1","WDR53","DMWD","SLC52A2","PIGP","IKZF1","PCYT2","NPAP1","BCAP31","ARL17A","GNB1L","DNAH14","CCDC190","EVI2B","NPIPB4","ZNF829","TMPRSS11B","THNSL1","TRIM69","ATP6V0C","IFITM1","LAMP1","POMK","C16orf54","KLHDC8B","SETD4","PTCH1","RTN4RL1","RNPC3","ZNF267","IRS2","C7orf61","FAM186A","SHOX","BICD2","CCIN","TMLHE","SLITRK2","PLK5","RASA3","LRCH3","NDUFA13","ZNF566","ZNF529","ZNF284","DLEU7","AIDA","C15orf41","CD300LF","ZPBP2","KRT5","GSAP","AGBL4","ARGFX","CYP2R1","LRRC70","ANKRD46","PIP5K1C","CYP4F2","ZBTB6","C2orf76","POLR3C","WWOX","CYP4Z1","CCDC84","BCL9L","POLR1D","KIF18B","ZNRF1","FFAR4","SAPCD2","EDARADD","SLC51B","CYP4F12","MARC1","SOWAHB","BLOC1S4","ZNF749","KLHL32","MRTFB","BTLA","ZNF17","KDM4D","GPAT2","TOR3A","PPP1CC","ZNF555","PRELID2","BACE1","TMEM212","SLC36A3","SLC36A2","THBS2","RXRA","ANKRD37","NUDT17","MINAR2","ZNF75D","CYP4X1","KRT10","GOLGA8R","CD300E","CCDC30","NKRF","GLDN","FCAR","KPNA4","ZNF501","ZNF197","SPATA12","TMPRSS12","RPS23","GNG2","BTN3A2","AKAP14","PCLO","RGS7BP","INSIG1","MYT1L","ZNF396","TMEM222","CLCNKA","ARHGAP30","SEPTIN10","FAM86B1","CYP4F8","CYP4F3","SMYD4","FOXD2","GPATCH8","CEACAM19","NF2","SMIM29","UBE2H","HPDL","KATNA1","FSD2","ARAP1","KIF24","PDE2A","SPDYE17","CARMIL3","PRR5","ZFP91","C17orf58","BCDIN3D","CYP27C1","LYRM7","CFAP73","CCDC73","BCR","OR10H1","FSCN2","FOXI2","SPIN4","SPIN2B","HYAL3","VSIG10L","CXCR3","ZNF397","ZSCAN30","TPCN1","LILRB4","TNFRSF4","KRT16","HEXIM1","KRT14","TRABD2A","PDZD7","POFUT2","MAPT","ERCC6L","TMEM17","TNFRSF18","ZDHHC17","ZNF395","ZACN","PPARA","TMEM232","FAM183A","EFCAB6","KANK3","RHD","ESPN","PNLIPRP1","PTRH1","GPR141","CYP4A11","TMEM216","RPS19BP1","TMPRSS11A","TMEM262","C3orf70","TEAD1","PLCD1","ENTPD5","MITF","HEATR4","NAP1L1","LILRA5","CMC1","SLIT1","LYPD6","AKR1C1","SPATA21","RNF220","ANGPTL5","SHTN1","LOC730098","ZNF546","TSPYL4","MT1X","GCNT1","SESTD1","FNBP1","DYNC2H1","KRT12","BCAM","RSBN1L","WDR86","EPOR","FAM9C","CIDEC","DCC","TAF9B","PCDHB13","MAGI2","LUZP2","CHP1","FPR3","KCNJ11","CDHR4","COL4A1","C12orf74","GJA4","PTMA","HSPA14","ATP13A5","SIRT7","PRR27","IFT140","TLR5","USP7","NHLRC1","DPPA3","PLEKHN1","ZNF385C","MAGEH1","TET3","ZNF286A","ISG15","EXD3","C17orf97","ZKSCAN4","RGPD1","DHRS4L2","PERM1","VMAC","HAPLN4","ERC2","B3GLCT","SPRY4","TRPV2","C2orf88","TMEM203","KBTBD12","THSD4","DNAJB13","GABRD","TCEA1","NHEJ1","FANCA","SECISBP2","C9orf153","SSX7","ADH1A","SEMA4D","DNAH17","MCRS1","FANCM","ZNF70","CARD9","PEAR1","ZFP69B","ZFP69","TMEM220","H1-2","PLSCR3","EIF4EBP1","OR6C75","CCDC157","FAM122A","PALM3","FYB2","LRRC74B","DMBT1","CLEC17A","LDLRAD2","C2orf66","CYHR1","COL14A1","DNER","KLHL17","PLA2G2C","ZSCAN23","RINL","C17orf99","OR7D2","TPRG1","MORN2","UBQLN2","RILPL1","C19orf67","ZNF490","NWD1","ARL4C","TMEM221","TREML4","RAB42","WNT7B","C11orf95","GPR89B","EYS","C6orf132","OR2AG2","MAPK12","NUTM2G","COL4A5","AGRN","NHS","FAM166A","TMPPE","ZNF626","HEPACAM2","SMTNL2","ZC3H6","LAMTOR4","PRKAR1B","NUTM2B","NCR3LG1","DCUN1D3","ZNF793","TUBB4B","AGAP4","COMMD6","IL17REL","HYKK","OR7A5","C15orf62","RUFY4","ZNF383","HES4","ZNF669","PEAK3","CENPP","PLSCR1","C3orf62","ENO4","ZNF559","SBK1","SLC38A3","GTF2F2","FAM92A","FOCAD","PRR19","ZP3","JAKMIP3","PDCD1","TCTEX1D4","SELL","CHM","BLOC1S5","CERKL","IER5L","H2AX","C19orf54","LCTL","NCCRP1","FAM83G","CFAP77","SRSF10","HBA2","DUSP28","CCDC9B","NBR1","OR2G6","RALGAPA2","NDOR1","CFAP54","CLN3","ASAH2","SUMO2","NANOS1","GOLGA8M","ZNF177","RTL6","DPYD","S100A16","PTAR1","CC2D2B","SAXO2","RHCE","IDO2","PARVB","SLC4A5","UROS","ZDHHC9","ZBED6CL","SMIM15","VWC2","FAM221A","TMEM120B","FSIP2","RBM34","NOXA1","TMEM198","BCL2L15","SPRED3","PLET1","ADRB3","PRELP","ZNF548","MTF1","SHISA6","TMEM201","NHLRC3","SNTN","ZDHHC11","CALHM6","CNR2","SLX4","ENTPD8","RPL14","ZNF563","TMC3","FBF1","ASTL","MSL1","LOC400499","LRRK2","GJB3","INSYN2A","TRMT2B","HACD4","CFAP126","FAM120AOS","UTS2B","C9orf152","NOC2L","MSANTD1","AADACL3","NELFB","ZNF292","KCTD21","SBSN","ADAT2","ZNF567","NDUFA4","ANKDD1B","ALKBH2","RNFT1","RNF222","RELN","FAM111B","APOD","H1-0","LITAF","TMEM120A","ARID2","SF3B3","PRSS48","BLOC1S3","SP6","PLAC9","NKAPL","CLDN4","ZNF573","FAM47E","JPT1","ZNF527","S100A13","ZNF33A","ZNF600","KIAA1107","MAOA","C15orf61","NUGGC","TSPYL1","PNRC2","DRICH1","GJB5","FHIT","ZKSCAN3","RRP7A","LIN54","FAM53B","S100A14","KAZN","SLC35E2B","TOGARAM2","NEMP2","ALG1L","KIAA0408","GSPT2","C8orf76","CXCL17","OTUD6A","HMGB1","SH2D5","NCR1","RASSF10","GJB4","BLOC1S2","SYCP2","ZNF724","IL1RAP","PTPRT","PAX5","ZNF676","ZNF699","TDRD7","CCDC189","OR8A1","KIAA0895L","HLA-DRB1","MYT1","SERPINA3","AKR1C3","SPATS2L","ZNF250","WDSUB1","ZNF79","S100A4","PLEKHG4","FAT4","KIF19","ZNF681","ACADSB","STK40","OR10J1","TMEM63A","SEMA4A","MPHOSPH8","GREB1","SIRPB2","ZNF766","RYR1","SRGAP3","FAM217B","TUBB","LCOR","SUPT5H","XPNPEP3","OR2C3","ZNF107","PPIA","ZNF471","ZNF836","ZNF493","GTF2IRD2","GRM7","SUPT3H","NIF3L1","ATP2A1","IARS1","MFSD14C","POM121","ZBTB44","GIMAP5","STK31","CGB7","NLGN3","OR8D1","ADH7","ZKSCAN7","CD55","ZNF565","NTNG2","ELAVL3","WDR5","LONP1","TRRAP","SRGAP2B","FUT4","ASB13","ZNF34","ZNF781","ZNF140","INCA1","ZNF774","PTPN1","EVL","THEM5","EPHB4","ZNF765","ZNF124","XRCC6","S100A5","C20orf204","PPP1R26","TSC22D2","NPIPB15","ZNF569","ARMCX4","YRDC","ZNF777","PIK3R4","ZNF775","ZNF605","TRAPPC2","MYL6B","ZNF799","SIAH1","C20orf96","ESRRG","IPO4","NCOR2","SULT1A1","ARL9","PRPF40A","GDAP2","TCEAL3","ANAPC7","TPK1","SLC6A9","AFAP1","NACA","MYO18A","SPTSSB","BORCS6","MAN2A2","MME","FAM72A","CCDC196","CACNA1H","SULF2","LAMA2","PLXNB2","AJAP1","XRCC2","MYO6","MRTFA","HDAC2","ZNF782","SLC22A25","ZNF846","MMP1","ADH1B","TCF4","WNK3","SDHAF3","HRH1","RABL6","ZNF136","ZKSCAN5","ZNF502","TRAPPC4","TTC30B","TECPR2","TLR7","FAM180B","ZFP62","ERI2","TOMM7","HSH2D","TRPV1","ZNF33B","ZNF512B","AMZ2","ZNF431","NF1","VKORC1L1","ZNF418","DAPK1","HLA-DQA1","COL27A1","GM2A","H2AC13","S100A2","ZNF700","CD47","OR52K1","TLE1","MAML3","H2AC11","STRN3","CHRNG","ZSCAN16","MVB12B","ILRUN",null,"OR11G2","POTEI","ADA","ARID5A","PATE2","PPTC7","KRT39","NHLRC2","H2AC7","ZFP28","KIAA1211L","CBWD3","SCN8A","LAMB3","H2BU1","KPNA5","ANKRD36B","ARHGEF12","HCAR1","PDLIM7","FLNA","SRGAP1","FAM3C","NOP9","SLC39A10","CASP4","AP2A1","ZNF585A","FUT11","ANXA4","LAGE3","WDR5B","NPIPB9","WDR45","METTL9","ZNF138","ZNF429","ZNF470","SERTAD1","ZNF100","CXorf40B","ZNF398","ZSCAN25","ANXA6","ZNF441","GMFB","SIGLEC15","ZNF420","ZNF763","ZMYM1","DTHD1","H4C3","ZSCAN26","MAFG","ARRDC1","KIAA1671","IGF2R","GAL3ST4","DYNC1H1","SLC6A17","PCBP2","ZGPAT","SLC25A29","PGAP1","SRC","ZNF682","OR8B8","ZNF772","ZNF257","PCNX3","ADAM32","ACSL5","LRRC8B","ABCB8","H3C12","SND1","ZNF785","SULT1A2","NEK5","PSMD12","PIWIL2","NOL4L","CYSRT1","SLC22A4","ENTPD4","C1D","TBC1D9B","H4C11","FAM110D","SERPINA1","TPSB2","KANK2","C6orf141","GTF2E2","IL27","RAD54B","ZNF165","SYNGAP1","FITM2","BLM","ZNF720","DDI2","SVIL","TRIM33","LRP10","PELI1","ZNF655","MRPL21","LYPD2","UAP1L1","FBXL22","ZNF786","ZNF517","ZNF675","SLC22A5","DACT3","ADARB1","ZNF860","HTT","C5AR1","CYP2B6","H3C4","DCHS2","VEPH1","SHPK","IPP","MAP3K5","CYP2F1","GSTK1","HNRNPAB","OR2L5","STMN3","PDGFA","GYPE","SPN","ZNF695","PCDHB11","ZNF628","SLC2A10","ZNF665","RPF2","SLC28A3","FAM177B","MIB2","OR6Y1","MYO5A","GZMM","ATG7","SIPA1L1","TTC30A","RAB40C","PIGN","COL4A6","ZNF624","HHLA3","TOPORS","BCO2","KCNMB2","ENTPD6","DMBX1","ENPP1","CCDC154","FAR1","CPLANE1","ZNF841","MFAP5","ZNF615","CXorf40A","CDC42SE1","MPEG1","SERPINB2","DPP4","SERPINB13","PDCD1LG2","ZNF433","DNAH10","SPTAN1","NMB","PARVA","KLHL14","FAM114A1","RPE","ZNF460","CR1L","PHF2","RPS26","C14orf178","PSAP","S100A10","CFAP43","LHFPL5","RPL37A","TXNRD3","CFD","STPG3","MCMBP","EME2","KLHDC1","ZNF81","TAF13","ZNF780A","ATAD3A","FAM118B","ZNF461","CCDC180","SLC9A8","OCLN","CFAP299","H4-16","CYP2A13","ZNF181","OR8G1","INKA2","ZNF44","GPAA1","ADAMTSL2","SGTB","ZNF790","PRB3","FAM49A","MYO1C","NKIRAS1","MEIG1","KIF13B","ADH5","H2BC12","SPG7","HRNR","ZNF677","ERO1A","ZNF823","ZNF311","ZNF347","PLCG2","FCHSD1","ZNF71","AADACL2","S100A6","RPL12","DNM3","ZNF121","MPZL1","VPS13A","MBP","AKAP17A","GOLGA6L9","LEKR1","C1orf122","CLEC9A","NOL8","IRAK4","CCDC151","MRPL42","ENTPD7","FCGR1B","ZNF335","RPS4X","ZNF273","ZNF84","MAK16","ZNF667","SIRPA","GRK6","PRIM1","MARCHF5","POTEH","NPIPB13","AKR1B10","SULT1C4","CYP2A7","ZBTB14","CD2AP","NUP62CL","SFI1","ZNF649","ADH4","OR2T6","ZNF248","TOR4A","LPAR1","MB","OR2L3","HIBCH","ZNF544","TMEM229B","SOWAHC","ZNF770","NPIPB6","HMGN5","MIER1","MAN1A2","SVIP","ZNF251","DDRGK1","TFDP1","CLEC4C","ZNF607","BPIFA1","ZNF334","HSD17B11","SZT2","SULT1C2","ZXDA","RPS6KL1","CACNA1E","QRICH1","CSF2RA","DDX42","RPL23A","SLC29A3","STYX","UBL5","HELZ","TMEM116","UCKL1","CARD11","ZNF485","SDAD1","ZKSCAN8","PHETA1","HYLS1","ZNF442","ZNF813","PIM3","GET3","ASPH","SPRED2","WWP2","GFPT1","UVRAG","ZNF26","ITSN2","NTRK1","OGA","MT1F","TCAF1","ZNF69","TXNRD1","NRARP","ZNF583","ZNF568","ZXDB","ZNF480","ZNF587","TPM2","RTP2","SH3BGRL2","ZNF808","ANKRD35","B3GNT6","YTHDF2","TMA16","HLA-DRB5","ATL1","CNGA1","MAFK","ARMH1","ZNF43","GPN1","PLN","C2CD4A","ZNF28","ZNF511","C20orf203","ZNF627","KCNRG","WDHD1","ZNF789","CTNND1","DDX39B","SLC34A3","RD3","SH2D1B","ARC","NUDT16","TLK1","LRBA","BAZ1A","AKR1C4","COPS8","CCDC69","MDM4","RYR2","KLHL9","FAM3D","NCOA6","STK39","TAT","C6orf89","CALM1","LPA","TAFA2","TTC37","TUSC1","PAPSS2","SLC9A6","FAN1","ABCA4","EIF1AY","ND6","IPO9","CEP290","COX2","GLMP","TOGARAM1","DLL1","ANKRD13B","ECI2","UNC13B","TEX45","CYTB","LDB1","PPP1R14C","CTR9","SMOC1","F5","MSRB1","ZNF652","SMURF1","SLC5A3","GPATCH3","CDC42BPB","PLXNB3","RPL10A","COLGALT2","EPS8L3","EGFL6","ND2","SYCP1","APCDD1L","RCSD1","RASSF9","FAM169A","ZNF830","GRIN3A","ND5","MUC2","CNOT7","TMEM184B","MTOR","SCAMP5","ALPK2","LRIG2","COX1","PNP","PAX9","LRRC10","GK","FOXJ3","ZNF358","SFT2D1","CD247","GRM3","CHAMP1","INPP5F","ARHGAP11A","SUCNR1","HMGN2","SELENOM","UBE2J1","GJC2","OPA1","DENND4B","RYR3","ZNF277","ND3","KTI12","SELENOT","TOX","CES1","CD3E","RUSC2","FICD","OSTC","R3HDM4","TSEN15","LTN1","RUNDC1","CCDC152","STKLD1","GRK5","TYW1","DCAF12","SFMBT2","ITPRIPL1","ND4","SMC5","ND1","PRMT6","SHISA4","CIPC","CAPZA2","ATP6","TOP1","PRC1","BHLHB9","MAP3K3","SREBF2","C1orf174","RASGEF1A","SPOUT1","RPL39","DZIP3","KIAA0753","DCLRE1A","ATG9A","NOS1AP","APRT","GPRASP1","TBKBP1","MAGEE1","CCDC167","COX3","ZFP2","SOWAHA","L3MBTL3","DMD","MFAP3L","NAGA","SMG5","KIFBP","TGM2","ARMCX6","PJA2","RORB","SGMS1","OR10Z1","ZNF525","INF2","FAM83H-AS1",null,"OR2L2","EFCAB2","COX20","CHML","CAPN8","TATDN3","CR1","RAET1G","SAMD5","TEDDM1","ECT2L","GPR52","FCGR3A","SPRN","FAM229B","FANK1","PRR9","SPRR2E","EEF1AKMT2","DDO","PLPP4","H3C14","H2AC18","H2BC18","H3C15","RBM20","C6orf163","GDI1","PCMTD2","LIME1","DPPA5","SAMD13","RTL8A","CCDC160","C1orf141","EFCAB7","LDLRAD1","RHOXF2B","ZYG11A","LIPN","LRRC73","FOXO6","SYS1","INPP5B","NEU4","MAFB","TRAF3IP1","CHIC1","GIGYF2","C2orf72","RUFY2","NHSL2","PHACTR4","ASAH2B","AGAP6","TIMM23B","ZDHHC18","TMEM273","CXorf65","AGAP9","SYT15","MACO1","PTPN20","GDF5OS","ZDBF2","TXNDC8","DAXX","BMPR2","TCEA3","PFDN6","RING1","HSD17B8","RXRB","OXLD1","HLA-DOA","BRD2","HLA-DMA","COL5A2","PSMB8","TAP2","SPIN3","NBDY","HLA-DRA","COL15A1","NOTCH4","PBX2","AGER","RNF5","AGPAT1","PJVK","PRRT1","FKBPL","MRPL38","SMIM5","ERICH2","STK19","CD300LD","DXO","SKIV2L","NELFE","ZBTB12","SDHD","EHMT2","LAYN","SLC44A4","NEU1","HSPA1B","HSPA1A","HSPA1L","LSM2","VARS1","VWA7","CARD16","MBD5","MSH5","MPIG6B","LY6G6C","ABHD16A","LY6G5C","CSNK2B","GPANK1","C6orf47","FAM155A","APOM","BAG6","PRRC2A","AIF1","LST1","NFKBIL1","ZNF814","MICB","ZNF551","MICA","ZNF805","HLA-C","POU5F1","CCHCR1","PSORS1C1","MUC21","DHX16","C6orf136","MRPS18B","PPP1R10","KRTAP5-11","ABCF1","PRR3","LILRB3","DDR1","GNL1","HLA-E","TRIM39","ZNF468","ZNF616","TRIM31","RNF39","PPP1R11","RACK1","HLA-G","TBC1D8","HLA-F","ASPDH","MOG","OR2H2","AKT1S1","GABBR1","C10orf113","OR11A1","OR14J1","OR2J3","C9orf135","TRIM27","RANBP17","INSYN2B","FOXD4L5",null,"TTC25","MRPL53","MROH6","ATXN2","DCTN1","TCTN1","FAM216A","ZBTB48","IGFL2","IGFL4","KRT40","MZT1","SPINK9","ZNF155","UQCC3","FBXO48","FAM221B","CD177","PSG5","ZNF783","ZNF425","LRRC10B","C12orf73","PCDHGA1","PCDHA9","PCDHA5","PCDHA4","TRIM13","SPIRE2","PKHD1L1","SLFN12L","SLC35B4","LGALS7","SYCE1L","TMEM231","FAM71F2","C2orf91","CCNI2","CDKL4","C4orf47","TRIQK","SDHAF1","PSENEN","C11orf91","ZBTB10","C4orf46","SCGB2B2","CCDC144NL","LGR4","PSMB10",null,"SPDYE2","E2F4","PDE7A","TMEM170B","MUC12","SNX2","SAP25","SARNP","ADGRG1","IPO7","PRR13","TECPR1","SLCO6A1","INSYN1","CFI","SAMD9","KRT6A","CNEP1R1","KRT81","EXOC3L4","ATP6AP1L","CCDC85C","C2CD4B","RGL3","NAP1L4","TMSB4X","TMEM256","CPT1B","SMN1","SERF1A","HMGN1","MUC19","DENND6B","EIF3CL","LCMT1","MFSD2B","CDPF1","HTN3","LIN52","SMIM11A","TECRL","DPF3","ETFRF1","C17orf107","ITSN1","ITPRIPL2","DENND1C","SLCO1B7","CRYZL1","C5orf51","GAGE1","PLPP6","KLRC2","GMNC","TTC23L","C1QTNF9B","ZNF316","ONECUT3","C21orf62","RNPS1","NYNRIN","DNAJC19","DEFA1","DOK6","JPT2","SERPINB11","SERPINB4","SERPINB5","ZDHHC11B","CFAP99","GOLGA8O","TMEM191C","HBA1","ELOA2","ATP10A","ANKUB1","FOXL2NB","COL6A6","RAB12","TMEM200C","HLA-A","HACD2","CFAP44","CD200R1L","LNP1","VGLL3","TRIM71","ZCWPW2","ANKRD28","COLQ","METTL6","XKR4","GPX3","DIO2","SELENOH","DIO1","STK38L","TSN","SLC48A1","TSSK1B","PRR22","TAS2R19","TAS2R14","TAS2R13","CTAGE1","RTL8B","OR2A42","RNF208","ND4L","MAP10","POU5F1B","ZNF580","ZNF611","NUP62","DENND1B","SFT2D2","FGFR1OP","SCAF8","CFAP45","ZNF254","TCTEX1D2","CRYGS","CRIP1","KLHL23","LINGO4","TRIM59","MLLT11","ASIC3","GIMAP1",null,"CCDC183","ARHGEF35","OR2F1","DNLZ","SUPT4H1","NRAS",null,"LTC4S","ANKRD39","QTRT1","CHUK","MXD3","GSTM2","COG8","ARHGAP19","HAUS7","LCAT","PTPRCAP","PVRIG","GPC2","SIPA1","ERV3-1","SYNJ2BP","ARL2","TTLL13P","GBP7","RBMXL1","SRA1","STIMATE","DNAJC9","C8orf82","VDAC1","ZBTB9","TMX2","HEXA","NDUFS3","LEPROT","LBH","ADAT3","PPP1CB","SULT1A4","GPSM3","LAT","NCKIPSD","ATF6B","TREX1","S1PR3","SLC35F6","CLIC1","DDAH2","RPS29","ZNF134","GTF2H4","DDX47","ZNF888","ZNF845","KLRK1","EMP2","KCTD11","UBD","PPM1N","SLC23A3","LTB4R","LTB4R2","DNASE1","MDP1","CSNK1E","IRF9","GALT","CLDN9","ITGA1","NUDT19","ZNF726","ZNF99","TAX1BP3","AP1G2","ZNF90","NAXD","BORCS8-MEF2B","GANC","TTLL3","REPIN1","MRPL23","ZNF891","SMIM7","FBXO16","TSPAN4","CPNE1","ARL16","WEE2","LYRM4","MYCBP","TMEM213","PRCD","ALG3","SH3D21","SMIM30","C19orf38","C17orf67","FIS1",null,"ANG","SCART1","COLCA2","SPDYE3","MBLAC1","SOGA3","NEURL1B","HAUS3","BBIP1","AS3MT","CCDC103","PLIN5","SEC14L6","NOTO","PPME1","STARD10","NUTM2D","CPEB1","EML6","ZNF727",null,"ZSWIM8","C10orf105","ARHGEF33","IFRD2","CAPN14","ZBED1","TOMM6","HNRNPUL2","CSKMT","MS4A4E","FER1L6","MTCP1","DCDC2C","SMTNL1","NPIPA8","ZSWIM7","ARHGEF28","TBC1D26","LRRC69","CRPPA","NPIPA7","RTL10","COL28A1","PHB2","TCP11X2","NEURL4","GRID2IP","UBXN2B","CBWD6","MUC5AC","FAM166B","PEX26","C5orf49","FASTKD5","GOLGA8B","HOMEZ","DDX3X","VPS16","MYL5","ZNF407","NPEPL1","RPL17-C18orf32","SIAH3","EFCAB8","RSC1A1","TMEM242","TMEM167B","FAM72D","TNFRSF25","SLC35E2A","TSTD1","RPTN","CYB5RL","C1orf167","TTC34","ATAD3C","IFI30","CCDC7","FNIP1","CKLF","PAM16","CDC14C","CEBPZOS","ZNF579","CELA3B","RNASEK","NBPF1","UMAD1","FAM228B","LINGO3","ZGLP1","VAMP2","OR6B1","C6orf226","PPP3R1","FANCG","OR2A5","AP4M1","C2orf16","OR2A12","CEBPD","ZNF816-ZNF321P","ARIH2OS","ZBED8","OR1C1","NPTXR","FAM200A","PPP2R2A","ZNF880","TRIM16","TIGD1","XKR9","SLC12A8","APOL6","FADS3","OR2A1","CCNL2","UBA52","PPT2","ZNF630","TIAF1","BTBD19","FAM185A","POTEM","DCDC2B","C10orf55","EXOSC6","VPS52","ZNF844","CKMT1A","TINCR","GDF1","HLA-DPB1","C1QTNF5","CPTP","PRR29","C4B","ATXN1L","SMIM13",null,"NPIPA3","NDUFAF8","TMEM233","PLEKHM1","CT62","MKRN2OS","C2CD4D","ZNF469","MCRIP1","SLC26A6","FAM229A","ERCC6","NOL7","CTAGE4","PIGBOS1","FTCDNL1","TEX22","OR52I2","CROCC2","TMEM185B","LOC102725191","HSBP1L1","SRRM5","PGAM4","C13orf42","ERVMER34-1","C14orf132","WDR46","ZNF717","KLLN","PARG","AKR1B15","SCAMP4","LTB","MRLN","POLR2J2","OR2M3","ATP8","ORM2","FAM174C","OST4","C2orf92","NUTM2E","FNDC10","PROB1","ARL17A","DHFR","SAPCD1","RPL41","ORM1","PATL2","ZNF492","ZNF688","PET100","GK3P","FAM90A26","IQCF3","ANKRD66","ACBD6","NFE4",null,"ANKRD18B","PPP5D1","CCDC192","PRB4","YY2","STMND1","HSBP1","OR5H1","CFAP97D1","HLA-DPA1","RPS18","TSPAN19","CYP21A2","PRH1","TAPBP","ZBED9","TMA7","TEX50","MCTS1","MTRNR2L4","OR52I1","SMIM26","AJM1",null,"HLA-DQB2","GOLGA8N","TNF","PET117","LYRM9",null,"RNF224",null,"BTBD18","TMEM238",null,"H2BC15","RPS28","UQCRHL","TRIM26","ZNF879","CCDC188","ZNF736","PINLYP","C5orf58","FAM133B","MCIDAS","JRK","NPIPB2","HLA-B","IQCM","HNRNPUL2-BSCL2","SHISA8","ANKRD65","BRD3OS","ZSCAN31","C12orf75","HGH1","PPP1R3E",null,"SPAAR","SMIM27","NFAM1","ANKRD34C","KIAA0040","B3GALT4","OR5H14","ZBTB22","ZBED5","PPIAL4G","CCDC200","UBE2L5","ZNF853","ARHGEF38","CTAGE9","B3GNT9","CDKN2AIPNL","CKMT1B","RNF223","PATE4","ZNF737","RGL2","C10orf143","SHISA9","OR7E24","HLA-DQA1","KIFC1","C2orf74","IRGM","AMY1A","FAM200B",null,"LRRC37A2","TMEM250","TXNDC5","CASTOR1","RNF103","RBM14","ALKBH6","ASB14","KLHL41","CASTOR3","STPG4","NME1","TNFSF12","CDRT4","APOBEC3G","TLR9","WBP1","MRPS17","GET4","C1orf226","ADSL",null,"LILRA2","TEX35","AMY2B","STRIT1","LY6G5B","PSMB9","PCDHGC3","SMKR1","UGT1A5","COX19","ACAD11","PPIL3","FOXO3B","TNFRSF13B","AQP1","C1QTNF9","ISY1","PNMA2","LRRD1","PCDHGC5","TMEM189","RDH14","PLCXD2","MIF","NSUN6","HLA-DOB","YAE1","TDGF1","C3orf85","CRCP","CDRT1","RPL36A","PDXP","RPP21","CD302","EGFL8","ATP5MF","ARHGAP8",null,"ARPC4","CORT","UGT1A1","INMT","ARPC1A","SPRR2A","ATP5PO","PLEKHO2","C8orf58","PISD","HOGA1","PWP2",null,"PI4KA","HYPK","AMACR","MTFP1","TCP10L","PSG2","ARFGAP3","C22orf39","UGT1A8","EIF6","PCDHGC4","MRPL20","ARPIN","UGT1A10","SERPINB10","HLA-DMB","DECR2","GNG10","CNTF","CCDC169","RTL5","AP5Z1","ZNF709","STRC","ERVW-1","EIF4EBP3","PSG11","UGT1A3","MRPL33","MICAL3","PPAN-P2RY11","PCDHAC2","STON1","PRAF2","VSIG8","STMP1","KCTD7","EFNA4","TICAM2","NAA80","TNFRSF6B","WFDC6","UPK3B","SMIM34A","IL10RB","CFB","ZNF487","WDR92","NME2",null,"PLA2G4B","CFAP57","NPIPB5","TTC4","TMEM35B","JMJD7","APOBEC3D","OR2A7","TUBA4B","MRPS6","ZNF512","GSTA1","ACY1","NFS1","DDOST","TMEM199","GSTA2","DNAJC25-GNG10","UGT1A7","P2RY11","TMEM141","TMEM225B","IFITM10","DBNDD2","ETV5","RBM12","UGT1A4","ERVFRD-1","LILRA6","SCARF2","APOBEC3C","CCDC13","ASPRV1","UBE2V1","CTAGE8","PTCHD4","C4A","HBB","CRYBB2","N4BP2L2","ZNF585B","CEBPA","H2AJ","UBAP1L","PGAM5","ZCCHC3","SPTY2D1OS","TWF2","MARS2","USP51","BCKDHA","INSL3","ADH1C","APELA","CDK11B","PCDHAC1","POU5F2","PCP4L1","ABHD14A","STIMATE-MUSTN1","RBM14-RBM4","LY75-CD302","TRIM59-IFT80","CCDC153","SMIM31","TNFSF12-TNFSF13","FMN1","HAUS5","EPPIN-WFDC6",null,"ATP5MGL",null,"TMEM150C",null,"NAIP","ZNF324B","SPATS1","IFNAR2","ZNF564","FER1L5",null,"RNF103-CHMP3","PDCD6","TERB1","TMEM158","ARPIN-AP3S2","YJEFN3","PCDHA10","ARPC4-TTLL3",null,"TRMT9B","ZNF718","SMIM20","TUNAR","CHCHD10","CDK3","GPR162","ATP6V1E2","GLI4",null,"REELD1","SELENOP","LOC100505841","ZNF674","TMED7-TICAM2","ZNF345","ALG1L2","SHANK3","ZNF550","CENPS-CORT",null,null,"PCDHGA12","SHLD3","CCDC71L","TMEM200B","PCDHGB6","SERPINE3","C1orf210","TRNP1","SMIM18","PCDHGA5","PCDHGA7","SLC10A5","EIF5AL1",null,"ALG11","ATXN7L3B","PRKDC","PCDHGA6","PCDHGA8","UTP14C","PCDHGA10","PCDHGA11","PCDHGB2","PCDHGB4","CLDN23","ZNF260","LYN","PINX1","PCDHGB7","CD8B2","NPIPB11","CHKB-CPT1B","SIGLEC14","PBOV1",null,"AP5B1","CHMP4A",null,null,"FPGT",null,"IGLL5","MEX3A",null,"OR10G4","EEF1G","SYS1-DBNDD2","SLC22A18AS","NPIPA2","MPV17L2","ATP6V1G2-DDX39B","BORCS8","INMT-MINDY4","DPP3","STX16-NPEPL1","ANKHD1-EIF4EBP3","BRK1","SAA2-SAA4",null,"CHMP1B","EID3","MSH5-SAPCD1","HTD2","NANOGP8","CARD17",null,"EID1",null,"NOX5","HCAR3","EBLN2","TUT1","POLR2M","FDXACB1","RAB44",null,"OR6J1",null,"MTRNR2L8","TIFAB","TAS2R20",null,"CYP2A6","SNHG28","CTSO","COA8","TRAPPC2B","DNAAF4","ZNF432","TAS2R30","ZNF10","ZNF486","SMIM3","HMBS","ZNF225",null,"TAS2R31","DND1","SALL3",null,"POLG2","SMIM10L1",null,null,"LIMS4","ZNF350","ZNF253","C17orf100","SLC5A8","LIMS3","HP","C11orf97","DENND11","LSM14A","NHLRC4","GATC","ZBED6","MGAM",null,"FNTB",null,"ST6GALNAC6","ZNF625","GALNT4","INAFM1","CNPY2","MGAM2",null,"CUX1","TEN1","P2RX5-TAX1BP3","MAP1LC3B2","CLEC5A","CHURC1",null,"C17orf49","RTEL1","PPT2-EGFL8","ZNF578","PDF",null,null,null,"SPESP1",null,"SPECC1L-ADORA2A","BCL2L2-PABPN1","SYNJ2BP-COX16",null,"TRIM34","PRORP","RNASE4","MC1R",null,"CEP95","TUBB3","UBE2F-SCLY",null,"TVP23C-CDRT4","FPGT-TNNI3K","BLOC1S5-TXNDC5","POC1B-GALNT4","NDUFC2-KCTD14","SMIM6",null,"ITGB3","SLC35G6","ZHX1-C8orf76",null,"INAFM2","ST20-MTHFS","CTXND1","THTPA","MRPL46","UBE2Q2L",null,null,"RBM15B","TGFBR3L",null,"SPDYE6","CCDC187","FRRS1L","PMF1-BGLAP","MRC1","SCX","C16orf95",null,"DUX4",null,"ANKRD20A1",null,null,"CCPG1","SULT1A3","TMEM178B","MPV17L",null,"EPPK1","CLEC19A","ZNF865","BOP1","MUC22","LOC105372440","PECAM1","TEN1-CDK3","TUBB8",null,"GAN",null,"GOLGA6L7","C15orf65",null,"GOLGA8S","BOLA2-SMG1P6","TCF24","GOLGA8H","ISY1-RAB43","CLN3","MIA",null,null,"PCDHGA9",null,"PCDHGB3","CORO7",null,null,"MMP12","SPDYE17",null,"SPON1","OVCA2","MRPL12","CCNQ","GTF2I","ZNF234",null,"MYZAP",null,"TMEM238L","SRSF8","IKBKE",null,"MSMB","CRHR1","LINC00672","NBPF11","RHEX","AKR1C8P",null,"ANXA8L1",null,"NOTCH2NLA","DYNLL2","OTUD7B",null,null,"ANXA8","RBM8A","TIMM23","RNF115","C21orf59-TCP10L","RPL17",null,"SEC22B","RAD54B","TXNIP","SRGAP2","BAHCC1",null,"RASSF5","STRADA",null,"NBPF15","NCOA4","MRPS21","MYO15B","FXYD1","AARSD1","ZNF850","PTGES3L",null,null,"C17orf113","NPFF",null,"UPK3BL1",null,null,"ZNF285","S1PR2","POLR2J2","FDX2","ZNF224",null,"EDDM13",null,"SMIM22","LIN37","NDUFA7","NBPF12","SLC6A14",null,"SMIM17","TCP11X1",null,"FAM156A",null,null,null,null,null,"ARHGEF18","ERVV-2","MTRNR2L2","FBXO17","TMSB15B",null,"MAGIX","IKBKG","ZNF587B","SPIB","NXF2","NXF2B","TMEM185A",null,null,"NBPF9","CCDC194","SLC25A53",null,"EGLN2","COMMD3-BMI1","FMC1-LUC7L2","ZNF559-ZNF177","TSNAX-DISC1",null,"NCBP2AS2","H4C15",null,null,"HEATR9","NBPF14","TAF15","GAS2L2","RPS10-NUDT3","H4C14","RASL10B","TLCD4-RWDD3","UGT2A2","SRXN1","NBPF19","NBPF10","MMP28","CCL5","PPIAL4E","LIX1L","MILR1","MROH7-TTC4",null,"ANKRD34A","GTF2H5","H2AC19",null,"NUDT3","KMT2B","POM121C","CD24",null,"FAM47E-STBD1",null,"CFAP206","ZNF595","DOC2B","PCDHB16","DCP1A","ATP6V1FNB",null,null,"C2orf15","GRIN2B","NBPF26",null,"SPATA13","SNURF","H3-2","ZBTB8B","C1QTNF3-AMACR",null,"SPDYE17","AGBL1","CWC25","SMIM11A","EPOP","ZNHIT3","H2BC14","CYFIP1","H2BC8","USP27X","TAF9","NOL12","GOLGA6L1","H3C8","CASTOR2","NATD1","SOCS7","CBSL","ADRA2B","H2BC6",null,"ZNF658","TMEM269","TBC1D3","RCC1L","RIMBP3B","H4C6","H2BC17","ELOA3","H3C6","TBC1D3B","NPHP3-ACAD11","PANO1","TBC1D3I",null,"H2AC12","ZNF280B","MLLT6","PPP4R3B","SYNRG","NUDT18","ZNF2","H4C13","CCL16","LENG9","CCL4","PRAG1","H3C11","CCL18","FCGBP","HNF1B","LOC102724159",null,"XKR5",null,null,"AATF","H2BC9","H3C1","CCL15","RBM27","PRH1-PRR4","RIMBP3","ARHGAP23","TUBGCP5","U2AF1L5","SIK1B","DUSP14","UHRF1","ORAI1","CCL4L2","CCL3L1",null,"H4C9","ANKRD20A3","PIK3R6","TADA2A","OR4E1","KCNE1B","PIP4K2B","H2AC14","CCL14","H2BC3","PCDHGB5","RAB7B","DACH1","H2AC16","GSTT4","H4C5","H2AC8","LOC102723996","TYW1B","F8A3","H4C4","PIGW","F8A1","H2BC7","PCGF2","FAM243B","SRCIN1","GPR179","MARCKS","ZNF670","PKD1L3","GPIHBP1",null,"CCL3","LOC105371267",null,"LOC102724488","H3C7","PSMB3","SRD5A2",null,"CISD3","RDM1","DDX52","ZNF8","LOC110117498-PIK3R3","SSTR3","PRICKLE4","MYO19","GGNBP2","ZNF229","H2AC4","DHRS11","ACACA","TMEM191B","NR2E3","H2BC10","C11orf98","MRM1","H4C1","GOLGA6L10","ELOA3B","H2AC17","H4C2","H3C10","MRPL45",null,"OR10A6","TSTD3","OR2T11","OR2AG1",null,"PPIAL4F","GVQW2","GATD3B","PCDH20",null,null,null,"CCDC163","PAGR1","MATR3",null,"TMEM272",null,null,null,"TMEM265",null,null,null,null,"SCHIP1",null,"C13orf46","ZNF724",null,null,null,"SMIM33","TEX52",null,"CFAP97D2","CNTNAP3C","FAM240A","FAM95C","LITAFD","PRRT1B","ZCWPW2","C1orf50","FAM169B","EXOC3L2","MYOCOS","VSIG10L2",null,"PARL",null,null,null,null,"TPTEP2-CSNK1E",null,"SLURP2","HSPA14","SCO2",null,"C2orf81","LIMS3-LOC440895",null,null,null,"LYNX1-SLURP2",null,null,null,null,null,null,null,"EEF1AKMT4","PDE8B","TBCE",null,null,"SMIM41","LOC729966",null,null,"CCDC39","SEPT5-GP1BB","ARHGAP11B",null,null,null,null,null,"UPK3BL2",null,"LOC112694756","TBCE",null,null,null,null,"CHN2",null,null,null,null,"ZNF280D","ATXN7","SLC35D2","ABCF2","COMMD7","POLR2J3","SOD2",null,"ZNF883","C4orf36",null,"ZNF365",null,null,null,null,null,null,null,null,"POLA2",null,null,null,null,"RNF43",null,null,null,null,"LRRC57","SPICE1","KRIT1",null,null,"RAET1E",null,"NOTCH2NLB",null,"GGT1",null,"ZDHHC11B","ZNF844","NOTCH2NLR","KYAT1",null,"CHTF8","AHRR",null,"LOC728392",null,"NOTCH2NLC","SPDYE11",null,"ZNF761","FKBP2","H3C2",null,"H3C3","HERC3","TPCN2",null,null,null,null,null],["tetraspanin 6","dolichyl-phosphate mannosyltransferase subunit 1, catalytic","SCY1 like pseudokinase 3","chromosome 1 open reading frame 112","FGR proto-oncogene, Src family tyrosine kinase","complement factor H","alpha-L-fucosidase 2","glutamate-cysteine ligase catalytic subunit","nuclear transcription factor Y subunit alpha","sperm tail PG-rich repeat containing 1","NIPA like domain containing 3","LAS1 like ribosome biogenesis factor","ectonucleotide pyrophosphatase/phosphodiesterase 4","semaphorin 3F","CF transmembrane conductance regulator","ankyrin repeat and IBR domain containing 1","cytochrome P450 family 51 subfamily A member 1","KRIT1 ankyrin repeat containing","RAD52 homolog, DNA repair protein","BCL2 associated agonist of cell death","leucine aminopeptidase 3","CD99 molecule (Xg blood group)","heparan sulfate-glucosamine 3-sulfotransferase 1","amine oxidase copper containing 1","HECT, C2 and WW domain containing E3 ubiquitin protein ligase 1","mitotic arrest deficient 1 like 1","LIM and SH3 protein 1","sorting nexin 11","transmembrane protein 176A","mannose-6-phosphate receptor, cation dependent","kelch like family member 13","islet cell autoantigen 1","dysbindin domain containing 1","alsin Rho guanine nucleotide exchange factor ALS2","caspase 10","CASP8 and FADD like apoptosis regulator","tissue factor pathway inhibitor","NADH:ubiquinone oxidoreductase complex assembly factor 7","RNA binding motif protein 5","myotubularin related protein 7","solute carrier family 7 member 2","ADP ribosylation factor 5","sterile alpha and TIR motif containing 1","DNA polymerase delta interacting protein 2","plexin D1","adenylate kinase 2","CD38 molecule","FKBP prolyl isomerase 4","lysine demethylase 1A","RNA binding motif protein 6","calcium/calmodulin dependent protein kinase kinase 1","RecQ like helicase","VPS50 subunit of EARP/GARPII complex","Rho GTPase activating protein 33","NADH:ubiquinone oxidoreductase subunit AB1","pyruvate dehydrogenase kinase 4","solute carrier family 22 member 16","zinc finger MYND-type containing 10","ATP binding cassette subfamily B member 5","aristaless related homeobox","solute carrier family 25 member 13","suppression of tumorigenicity 7","cell division cycle 27","solute carrier family 4 member 1 (Diego blood group)","holocytochrome c synthase","dishevelled segment polarity protein 2","serine protease 22","UPF1 RNA helicase and ATPase","src kinase associated phosphoprotein 2","solute carrier family 25 member 5","mitochondrial calcium uniporter dominant negative beta subunit","RNA polymerase II subunit J","DEAH-box helicase 33","thrombospondin type 1 domain containing 7A","DNA ligase 3","RNA polymerase II associated protein 3","acyl-CoA synthetase medium chain family member 3","RNA exonuclease 5","cytokine induced apoptosis inhibitor 1","signal peptide peptidase like 2B","family with sequence similarity 214 member B","coatomer protein complex subunit zeta 2","protein kinase cAMP-dependent type II regulatory subunit beta","MSL complex subunit 3","CREB binding protein","TSPO associated protein 1","myeloperoxidase","GC-rich sequence DNA-binding factor 2","WD repeat domain 54","carnitine O-octanoyltransferase","ATP binding cassette subfamily B member 4","lysine methyltransferase 2E","rhomboid domain containing 2","inhibitor of Bruton tyrosine kinase","zinc finger protein 195","MYC binding protein 2","F-box and leucine rich repeat protein 3","integrin subunit alpha L","pyruvate dehydrogenase kinase 2","integrin subunit alpha 3","zinc finger protein X-linked","lysosomal associated membrane protein 2","integrin subunit alpha 2b","ankyrin repeat and SOCS box containing 4","glycerophosphodiester phosphodiesterase 1","required for excision 1-B domain containing","cytokine receptor like factor 1","oxysterol binding protein like 7","transmembrane protein 98","Y-box binding protein 2","mitogen-activated protein kinase kinase kinase 14","ATP binding cassette subfamily C member 8","transmembrane protein 132A","adaptor related protein complex 2 subunit beta 1","zinc finger protein 263","C-X3-C motif chemokine ligand 1","spermatogenesis associated 20","calcium voltage-gated channel subunit alpha1 G","TNF receptor superfamily member 12A","distal-less homeobox 6","mitogen-activated protein kinase kinase kinase 9","RAS like proto-oncogene A","BAR/IMD domain containing adaptor protein 2 like 1","lysine demethylase 7A","ETS variant transcription factor 1","acylglycerol kinase","aldehyde dehydrogenase 3 family member B1","tetratricopeptide repeat domain 22","putative homeodomain transcription factor 2","FERM, ARH/RhoGEF and pleckstrin domain protein 2","USH1 protein network component harmonin","gamma-glutamylcyclotransferase","DBF4 zinc finger","thromboxane A2 receptor","interferon related developmental regulator 1","galectin 14","cytochrome c oxidase assembly factor heme A:farnesyltransferase COX10","GTF2I repeat domain containing 1","PAF1 homolog, Paf1/RNA polymerase II complex component","VPS41 subunit of HOPS complex","Rho GTPase activating protein 44","elaC ribonuclease Z 2","scinderin","arylsulfatase D","patatin like phospholipase domain containing 4","adiponectin receptor 2","cyclin dependent kinase like 3","uridine phosphorylase 2","microtubule affinity regulating kinase 4","prominin 1","coiled-coil domain containing 124","CEA cell adhesion molecule 21","platelet activating factor acetylhydrolase 1b regulatory subunit 1","nitric oxide synthase 2","dynein axonemal heavy chain 9","KIAA0100","solute carrier family 13 member 2","growth arrest specific 7","trafficking protein particle complex 6A","megakaryocyte-associated tyrosine kinase","CEA cell adhesion molecule 7","suppression of tumorigenicity 7 like","transketolase like 1","paired box 6","RNA pseudouridine synthase domain containing 1","rhomboid 5 homolog 1","LUC7 like","calcium voltage-gated channel auxiliary subunit alpha2delta 2","BAI1 associated protein 3","TSR3 ribosome maturation factor","phosphatidylinositol glycan anchor biosynthesis class Q","cramped chromatin regulator homolog 1","TEA domain transcription factor 3","DnaJ heat shock protein family (Hsp40) member C11","flavin containing dimethylaniline monoxygenase 3","myosin regulatory light chain interacting protein","NADPH oxidase 1","E2F transcription factor 2","proteasome 20S subunit beta 1","jumonji and AT-rich interaction domain containing 2","cyclin dependent kinase like 5","calcium/calmodulin dependent protein kinase IG","cyclin dependent kinase 11A","NAD kinase","DLEC1 cilia and flagella associated protein","cytohesin 3","ADAM metallopeptidase domain 22","synaptophysin like 1","cytochrome b561","sperm associated antigen 9","cadherin EGF LAG seven-pass G-type receptor 3","aminoadipate-semialdehyde synthase","pleckstrin homology and RhoGEF domain containing G6","SS18 like 2","MPN domain containing","microsomal glutathione S-transferase 1","cryptochrome circadian regulator 1","peptidoglycan recognition protein 1","nuclear factor I X","ST3 beta-galactoside alpha-2,3-sialyltransferase 1","matrix metallopeptidase 25","interleukin 32","polycystin 1, transient receptor potential channel interacting","mitogen-activated protein kinase 8 interacting protein 2","mediator complex subunit 24","Rho related BTB domain containing 2","HEAT repeat containing 5B","SEC62 homolog, preprotein translocation factor","ribosomal protein S20","cold shock domain containing E1","ubiquitin protein ligase E3C","REV3 like, DNA directed polymerase zeta catalytic subunit","teneurin transmembrane protein 1","paired box 7","mannan binding lectin serine peptidase 2","iodotyrosine deiodinase","family with sequence similarity 76 member A","TRAF3 interacting protein 3","protein O-mannosyltransferase 2","vesicle trafficking 1","MLX interacting protein like","bromodomain adjacent to zinc finger domain 1B","RAN binding protein 9","ETS variant transcription factor 7","SprT-like N-terminal domain","eEF1A lysine and N-terminal methyltransferase","dual specificity tyrosine phosphorylation regulated kinase 4","zinc finger protein 207","ubiquinol-cytochrome c reductase core protein 1","STARD3 N-terminal like","CD9 molecule","hedgehog acyltransferase like","non-SMC condensin I complex subunit D2","intermediate filament family orphan 1","gastric inhibitory polypeptide receptor","PHD finger protein 7","nischarin","stabilin 1","fuzzy planar cell polarity protein","solute carrier family 6 member 13","iduronate 2-sulfatase","zinc finger protein 200","CD4 molecule","leucine rich repeat containing 23","Bruton tyrosine kinase","homeostatic iron regulator","Scm polycomb group protein homolog 1","FYN proto-oncogene, Src family tyrosine kinase","HIVEP zinc finger 2","flavin containing dimethylaniline monoxygenase 1","elongin A","lysophospholipase 2","chloride voltage-gated channel 6","mannose receptor C type 2","tetraspanin 9","BTB domain containing 7","amyloid beta precursor protein binding family A member 3","MKS transition zone complex subunit 1","abhydrolase domain containing 5","anosmin 1","A-kinase anchoring protein 8 like","mbt domain containing 1","UTP18 small subunit processome component","ring finger protein 216","tetratricopeptide repeat domain 19","polypyrimidine tract binding protein 1","double PHD fingers 1","synaptotagmin 7","leucyl-tRNA synthetase 2, mitochondrial","phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 alpha","plasminogen activator, urokinase receptor","anillin actin binding protein","WIZ zinc finger","G protein-coupled receptor 21","decorin","glutaminyl-peptide cyclotransferase like","protein phosphatase 5 catalytic subunit","centrosomal protein 68","mitogen-activated protein kinase kinase kinase kinase 3","zinc finger and BTB domain containing 32","TYRO protein tyrosine kinase binding protein","transmembrane protein 159","BRCA1 DNA repair associated","ERCC excision repair 1, endonuclease non-catalytic subunit","CD22 molecule","semaphorin 3B","membrane bound transcription factor peptidase, site 2","prickle planar cell polarity protein 3","lactotransferrin","exostosin like glycosyltransferase 3","ELOVL fatty acid elongase 5","arachidonate 5-lipoxygenase","lysine demethylase 5D","calcium binding and coiled-coil domain 1","ubiquitin protein ligase E3 component n-recognin 7 (putative)","mitogen-activated protein kinase kinase kinase kinase 5","EH domain containing 3","proteasome 26S subunit, ATPase 4","mannosidase alpha class 2B member 2","solute carrier family 7 member 14","claudin 11","solute carrier family 25 member 39","major vault protein","negative regulator of ubiquitin like proteins 1","phosphoglucomutase 3","RWD domain containing 2A","CDC like kinase 1","RNA polymerase III subunit B","angel homolog 1","ring finger protein 14","deoxyribonuclease 1 like 1","DEAD/H-box helicase 11","heme binding protein 1","G protein-coupled receptor class C group 5 member A","mastermind like domain containing 1","CD6 molecule","transforming acidic coiled-coil containing protein 3","UFM1 specific ligase 1","DNA polymerase alpha 2, accessory subunit","zinc finger CCCH-type containing 3","calpain 1","acid phosphatase, prostate","solute carrier family 30 member 9","myotubularin related protein 11","cytochrome c oxidase assembly homolog COX15","coiled-coil domain containing 88C","YY1 associated factor 2","zinc finger MYND-type containing 11","WASP actin nucleation promoting factor","BH3 interacting domain death agonist","matrin 3","xylosyltransferase 2","RANBP2 like and GRIP domain containing 5","stathmin 4","NudC domain containing 3","ISL LIM homeobox 1","choline dehydrogenase","interleukin 20 receptor subunit alpha","chloride channel accessory 4","glycosyltransferase 8 domain containing 1","ATPase secretory pathway Ca2+ transporting 1","insulin like growth factor 1","solute carrier family 38 member 5","ralA binding protein 1","RUN and FYVE domain containing 3","contactin 1","solute carrier family 11 member 1","WW domain containing transcription regulator 1","alkylglycerone phosphate synthase","chromosome X open reading frame 56","tetratricopeptide repeat domain 27","zinc finger protein 582","V-set and immunoglobulin domain containing 2","pleckstrin homology like domain family B member 1","cytochrome P450 family 24 subfamily A member 1","PR/SET domain 11","synaptotagmin 13","snail family transcriptional repressor 2","CD74 molecule","hepatocyte growth factor","zinc finger RANBP2-type containing 1","neurochondrin","adhesion G protein-coupled receptor A2","ZFP64 zinc finger protein","MNAT1 component of CDK activating kinase","sterile alpha motif domain containing 4A","RUNX family transcription factor 3","MRE11 homolog, double strand break repair nuclease","pleckstrin homology domain containing B1","serpin family B member 1","cytochrome P450 family 3 subfamily A member 43","solute carrier family 7 member 9","spastin","neurexin 3","oxysterol binding protein like 5","aquarius intron-binding spliceosomal factor","carbamoyl-phosphate synthase 1","four and a half LIM domains 1","replication termination factor 2","gamma-aminobutyric acid type A receptor alpha1 subunit","NLR family pyrin domain containing 2","solute carrier family 45 member 4","ring finger protein 10","zinc finger protein 839","zinc finger DHHC-type containing 6","GRAM domain containing 1B","ribonuclease/angiogenin inhibitor 1","NADH:ubiquinone oxidoreductase core subunit S1","RB1 inducible coiled-coil 1","endoplasmic reticulum protein 44","5'-aminolevulinate synthase 1","baculoviral IAP repeat containing 3","A-kinase anchoring protein 11","glutaredoxin 2","small nuclear RNA activating complex polypeptide 1","deoxyribose-phosphate aldolase","serine/threonine kinase receptor associated protein","ATP binding cassette subfamily C member 2","DEF6 guanine nucleotide exchange factor","pleckstrin homology domain containing O1","glutamate-cysteine ligase modifier subunit","ubiquitin protein ligase E3 component n-recognin 2","EH domain containing 2","DEP domain containing 1","coiled-coil domain containing 28A","Ras related GTP binding D","heat shock transcription factor 2","PHD finger protein 20","nuclear receptor subfamily 1 group H member 3","thymidine phosphorylase","non-SMC condensin II complex subunit H2","translocase of outer mitochondrial membrane 34","SEC63 homolog, protein translocation regulator","karyopherin subunit alpha 6","vimentin","RTEL1-TNFRSF6B readthrough (NMD candidate)","Fas cell surface death receptor","ribonuclease T2","CD44 molecule (Indian blood group)","potassium voltage-gated channel modifier subfamily G member 1","1-acylglycerol-3-phosphate O-acyltransferase 4","SLAM family member 7","butyrophilin subfamily 3 member A1","mitochondrial intermediate peptidase","protein kinase C eta","interferon gamma receptor 1","beta-1,4-galactosyltransferase 7","SH2 domain containing 2A","VRK serine/threonine kinase 2","TNF receptor superfamily member 1B","vezatin, adherens junctions transmembrane protein","POU class 2 homeobox 2","bromodomain containing 9","sorting nexin 1","TATA-box binding protein like 1","aryl hydrocarbon receptor nuclear translocator like 2","BCL2 associated transcription factor 1","solute carrier family 39 member 9","ankyrin 1","transcription factor B1, mitochondrial","rabaptin, RAB GTPase binding effector protein 1","high mobility group box 3","nucleoporin 160","BCL2 antagonist/killer 1","muscle associated receptor tyrosine kinase","IKAROS family zinc finger 2","granulin precursor","family with sequence similarity 13 member B","Rho GTPase activating protein 31","centromere protein Q","seryl-tRNA synthetase 1","RAN binding protein 3","AT-rich interaction domain 4A","EARP complex and GARP complex interacting protein 1","patatin like phospholipase domain containing 6","intraflagellar transport 88","ALG1 chitobiosyldiphosphodolichol beta-mannosyltransferase","zinc finger CCHC-type containing 8","ATP binding cassette subfamily F member 2","chondroitin polymerizing factor 2","leucine rich repeat containing 7","fucosyltransferase 8","ubiquitin like modifier activating enzyme 6","GRB2 associated binding protein 2","ATPase H+ transporting V0 subunit a1","protein inhibitor of activated STAT 1","solute carrier family 4 member 7","amyloid beta precursor protein binding family A member 2","mitogen-activated protein kinase kinase 3","EF-hand calcium binding domain 1","thymosin beta 10","asteroid homolog 1","ring finger protein 19A, RBR E3 ubiquitin protein ligase","peroxisomal biogenesis factor 3","GABA type A receptor associated protein like 2","SH3 and SYLF domain containing 1","family with sequence similarity 136 member A","vinculin","DEP domain containing 1B","death associated protein kinase 2","neutral sphingomyelinase activation associated factor","adenylosuccinate synthase 2","signal transducing adaptor family member 1","TIMP metallopeptidase inhibitor 2","replication factor C subunit 1","TBC1 domain family member 23","cullin 3","myomesin 2","cytochrome P450 family 46 subfamily A member 1","zinc finger ZZ-type containing 3","solute carrier family 18 member A1","ubiquitin specific peptidase 2","calcium sensing receptor","tubulin gamma 2","ribosomal protein L26 like 1","fms related tyrosine kinase 4","NOP2/Sun RNA methyltransferase 2","F-box protein 42","microfibril associated protein 3","methylthioribose-1-phosphate isomerase 1","methyltransferase like 1","aspartylglucosaminidase","phosphatidylinositol 4-kinase type 2 beta","biorientation of chromosomes in cell division 1 like 1","methionine adenosyltransferase 2B","tolloid like 1","enhancer of mRNA decapping 4","trio Rho guanine nucleotide exchange factor","versican","C-type lectin domain containing 16A","macrophage scavenger receptor 1","cadherin 1","Mtr4 exosome RNA helicase","dynein axonemal heavy chain 5","zinc finger FYVE-type containing 16","RHO family interacting cell polarization regulator 1","retinoic acid induced 14","polynucleotide kinase 3'-phosphatase","bestrophin 2","PH domain and leucine rich repeat protein phosphatase 2","spindle apparatus coiled-coil protein 1","staufen double-stranded RNA binding protein 2","solute carrier family 66 member 1","cystinosin, lysosomal cystine transporter","PHD finger protein 23","inositol polyphosphate-4-phosphatase type I A","RAB27B, member RAS oncogene family","proteasome 20S subunit alpha 4","myosin XVI","large 60S subunit nuclear export GTPase 1","poly(ADP-ribose) polymerase family member 3","tenascin C","THAP domain containing 3","RIPOR family member 3","tyrosyl-DNA phosphodiesterase 1","apoptosis inducing factor mitochondria associated 2","chromosome 2 open reading frame 83","spermatogenesis associated 7","mediator complex subunit 17","retinol saturase","capping actin protein, gelsolin like","adaptor related protein complex 2 subunit sigma 1","usherin","thyroglobulin","ADAM metallopeptidase domain 28","BARX homeobox 2","defective in cullin neddylation 1 domain containing 1","jade family PHD finger 2","lymphocyte cytosolic protein 2","tRNA isopentenyltransferase 1","adrenoceptor beta 1","cullin 7","catenin alpha 1","phosphorylase kinase regulatory subunit alpha 2","centlein","EPH receptor A3","heat shock protein family A (Hsp70) member 5","desmoglein 2","gem nuclear organelle associated protein 8","OFD1 centriole and centriolar satellite protein","glycoprotein M6B","phosphatidylinositol-3,4,5-trisphosphate dependent Rac exchange factor 2","WD repeat domain 37","YTH domain containing 2","CTP synthase 2","ATPase H+ transporting V1 subunit H","RNA polymerase II subunit B","family with sequence similarity 214 member A","ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 2","translocated promoter region, nuclear basket protein","ceruloplasmin","KIAA0556","dystrobrevin binding protein 1","X-linked Kx blood group","anoctamin 2","chromosome 12 open reading frame 4","Scm polycomb group protein like 1","WWC family member 3","Rho GTPase activating protein 6","family with sequence similarity 184 member B","microtubule associated protein 4","golgi associated PDZ and coiled-coil motif containing","ubiquitin specific peptidase 28","histone deacetylase 9","tetraspanin 17","NOP16 nucleolar protein","coiled-coil and C2 domain containing 2A","ribonucleotide reductase regulatory TP53 inducible subunit M2B","zinc finger protein 800","sorting nexin 29","LIM domain only 3","mitochondrial ribosomal protein S10","remodeling and spacing factor 1","vacuolar protein sorting 13 homolog D","CUGBP Elav-like family member 2","family with sequence similarity 120A","R3H domain containing 1","collagen type IX alpha 2 chain","KIT ligand","ERCC excision repair 8, CSA ubiquitin ligase complex subunit","ADAM metallopeptidase with thrombospondin type 1 motif 6","hexose-6-phosphate dehydrogenase/glucose 1-dehydrogenase","vesicle associated membrane protein 3","period circadian regulator 3","TNF receptor superfamily member 9","epsin 3","latent transforming growth factor beta binding protein 1","reticulocalbin 1","elastin","replication factor C subunit 2","AT-rich interaction domain 1B","CLPTM1 like","NEDD4 like E3 ubiquitin protein ligase","forkhead box P3","protein phosphatase 1 regulatory subunit 3F","hexosaminidase subunit beta","pentatricopeptide repeat domain 2","neurite extension and migration factor","JNK1/MAPK8 associated membrane protein","dickkopf WNT signaling pathway inhibitor 3","Rho guanine nucleotide exchange factor 5","nuclear factor, erythroid 2 like 3","mitochondrial calcium uniporter regulator 1","LIM domain and actin binding 1","LETM1 domain containing 1","solute carrier family 4 member 8","prostaglandin E receptor 3","TNFAIP3 interacting protein 3","mitogen-activated protein kinase 9","collagen type XXIII alpha 1 chain","BCAR1 scaffold protein, Cas family member","family with sequence similarity 160 member A2","homocysteine inducible ER protein with ubiquitin like domain 1","homer scaffold protein 3","RAD51 recombinase","DNA polymerase theta","phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta","cytochrome b-245 alpha chain","THO complex 3","heme binding protein 2","M-phase phosphoprotein 9","pleckstrin homology domain containing A5","serine protease 8","suppressor of IKBKE 1","ribosomal RNA processing 12 homolog","folliculin interacting protein 2","methylsterol monooxygenase 1","tetratricopeptide repeat domain 17","follistatin like 4","forkhead box N3","aldo-keto reductase family 7 member A2","MRT4 homolog, ribosome maturation factor","neuronatin","unconventional SNARE in the ER 1","MCF.2 cell line derived transforming sequence-like 2","nuclear receptor interacting protein 2","laminin subunit alpha 3","adaptor related protein complex 5 subunit mu 1","anaphase promoting complex subunit 4","potassium voltage-gated channel subfamily Q member 1","trafficking protein particle complex 3","thyroid hormone receptor associated protein 3","phosphohistidine phosphatase 1","ectonucleoside triphosphate diphosphohydrolase 2","lymphocyte antigen 75","AT-rich interaction domain 4B","opsin 3","serologically defined colon cancer antigen 8","hedgehog acyltransferase","kinesin family member 1B","forkhead box C1","TBC1 domain family member 22A","spectrin repeat containing nuclear envelope protein 2","pleckstrin homology, MyTH4 and FERM domain containing H1","ATPase phospholipid transporting 9A (putative)","family with sequence similarity 168 member A","RELT TNF receptor","galactosylceramidase","NOP58 ribonucleoprotein","SUZ RNA binding domain containing 1","cullin 1","family with sequence similarity 114 member A2","cytoplasmic FMR1 interacting protein 2","TGF-beta activated kinase 1 (MAP3K7) binding protein 2","glycoprotein integral membrane 1","eukaryotic translation initiation factor 2 alpha kinase 2","ubiquitin specific peptidase 36","lysine methyltransferase 2C","mucolipin 3","pumilio RNA binding family member 2","mitochondrial ribosomal protein L43","inter-alpha-trypsin inhibitor heavy chain 4","inter-alpha-trypsin inhibitor heavy chain 1","histone PARylation factor 1","zinc finger RNA binding protein","zinc finger protein 280C","TNF receptor associated factor 1","ring finger and CCCH-type domains 2","interleukin 17 receptor B","TRAF3 interacting protein 2","glycogenin 2","discoidin, CUB and LCCL domain containing 2","serpin family B member 3","sterol O-acyltransferase 1","plakophilin 2","GDP dissociation inhibitor 2","PR/SET domain 1","autophagy related 5","transmembrane and coiled-coil domain family 3","PITH domain containing 1","metastasis associated 1 family member 3","ubiquitin specific peptidase 13","ATPase phospholipid transporting 11B (putative)","laminin subunit gamma 2","cyclin dependent kinase 14","SEC61 translocon alpha 1 subunit","protein phosphatase 1 regulatory subunit 12A","Ras protein specific guanine nucleotide releasing factor 1","calcium/calmodulin dependent protein kinase II beta","ciliary rootlet coiled-coil, rootletin","RNA polymerase III subunit E","ATPase plasma membrane Ca2+ transporting 4","zinc finger CCCH-type containing 11A","RIO kinase 2","Yip1 domain family member 1","NDC1 transmembrane nucleoporin","diacylglycerol kinase gamma","FLYWCH-type zinc finger 1","unk like zinc finger","thromboxane A synthase 1","poly(ADP-ribose) polymerase family member 12","aldehyde dehydrogenase 18 family member A1","TAR (HIV-1) RNA binding protein 1","glutamyl-tRNA amidotransferase subunit B","MAX dimerization protein 1","cyclin dependent kinase 17","DnaJ heat shock protein family (Hsp40) member C25","solute carrier family 2 member 3","pleckstrin and Sec7 domain containing","CTD phosphatase subunit 1","Y-box binding protein 3","serine/threonine/tyrosine kinase 1","WNK lysine deficient protein kinase 1","cell division cycle and apoptosis regulator 1","opioid growth factor receptor","G protein subunit alpha 15","phosphatidylinositol glycan anchor biosynthesis class V","protein tyrosine phosphatase receptor type U","small nuclear ribonucleoprotein U5 subunit 40","RIMS binding protein 2","glutamine and serine rich 1","mitochondrial pyruvate carrier 1","acetyl-CoA acyltransferase 1","branched chain amino acid transaminase 1","histone deacetylase 7","leucine zipper tumor suppressor 1","PR/SET domain 6","sperm associated antigen 4","NCK associated protein 1","mitochondrial ribosomal protein S35","guanylate cyclase 1 soluble subunit beta 1","splicing factor SWAP","tyrosine kinase non receptor 2","MON2 homolog, regulator of endosome-to-Golgi trafficking","cadherin 3","GC-rich promoter binding protein 1","diacylglycerol O-acyltransferase 2","zinc finger protein 112","citrate synthase","leukocyte receptor tyrosine kinase","mitochondrial ribosomal protein S24","engulfment and cell motility 2","WAPL cohesin release factor","vacuole membrane protein 1","amyloid beta precursor protein binding protein 2","DNA polymerase delta 1, catalytic subunit","seizure related 6 homolog","eukaryotic translation initiation factor 4B","solute carrier family 6 member 16","BRD4 interacting chromatin remodeling complex associated protein","sphingosine kinase 2","ribosomal protein L18","carbonic anhydrase 11","isochorismatase domain containing 2","U2 small nuclear RNA auxiliary factor 2","epsin 1","mediator complex subunit 29","zinc finger protein 275","myotubularin related protein 1","glypican 1","aarF domain containing kinase 1","hydroxyacylglutathione hydrolase","ring finger protein 4","caspase 8","LIM and calponin homology domains 1","integrator complex subunit 13","transmembrane 7 superfamily member 3","sperm autoantigenic protein 17","tetraspanin 32","cellular communication network factor 5","doublesex and mab-3 related transcription factor 3","ST3 beta-galactoside alpha-2,3-sialyltransferase 6","ATPase secretory pathway Ca2+ transporting 2","nerve growth factor receptor","cell adhesion associated, oncogene regulated","TATA-box binding protein associated factor 2","homeodomain interacting protein kinase 2","transportin 3",null,"regulatory factor X associated ankyrin containing protein","transmembrane protein 161A","lysophosphatidic acid receptor 2","cathepsin A","SURP and G-patch domain containing 2","solute carrier family 12 member 2","sorting nexin 24","EYA transcriptional coactivator and phosphatase 2","calponin 2","ATP binding cassette subfamily A member 7","synuclein alpha interacting protein","DEAD-box helicase 20","BTB domain containing 1","fatty acyl-CoA reductase 2","breast carcinoma amplified sequence 1","chitinase 3 like 2","strawberry notch homolog 2","PMS1 homolog 1, mismatch repair system component","high mobility group 20B","calcitonin receptor like receptor","TATA-box binding protein associated factor 11","ankyrin repeat and sterile alpha motif domain containing 1A","adaptor related protein complex 3 subunit delta 1","zinc finger protein 76","SLC9A3 regulator 2","nth like DNA glycosylase 1","UHRF1 binding protein 1","G protein subunit alpha i3","importin 5","ornithine aminotransferase","WD repeat domain 3","protein kinase N2","WD repeat domain 18","translocation associated membrane protein 2","netrin 1","minichromosome maintenance 10 replication initiation factor","diacylglycerol kinase alpha","erb-b2 receptor tyrosine kinase 3","ankyrin repeat domain 44","lysyl-tRNA synthetase 1","adenosine deaminase tRNA specific 1","protein disulfide isomerase family A member 5","TBC1 domain family member 22B","NADH:ubiquinone oxidoreductase subunit B4","spen family transcriptional repressor","myosin light chain kinase","zinc finger CCCH-type containing 15","mitogen-activated protein kinase kinase 4","proton activated chloride channel 1","STE20 like kinase","cytochrome b5 reductase 4","collagen type XVII alpha 1 chain","glutathione S-transferase omega 2","SEC61 translocon alpha 2 subunit","protein kinase C theta","TLE family member 2, transcriptional corepressor","ankyrin repeat and SOCS box containing 1","family with sequence similarity 107 member B","malic enzyme 1","TBC1 domain family member 1","cyclin dependent kinase 13","methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2, methenyltetrahydrofolate cyclohydrolase","solute carrier family 9 member A7","forkhead box J2","Y-box binding protein 1","phosphodiesterase 4A","protein phosphatase 2 regulatory subunit B'alpha","catenin alpha 2","ELAV like RNA binding protein 1","tyrosine kinase with immunoglobulin like and EGF like domains 1","disco interacting protein 2 homolog B","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 1","lysine demethylase 4A","nuclear transcription factor Y subunit gamma","zinc finger MYND-type containing 12","solute carrier family 9 member A3","neuronal guanine nucleotide exchange factor","abnormal spindle microtubule assembly","CD84 molecule","ELOVL fatty acid elongase 1","Spi-1 proto-oncogene","zinc ribbon domain containing 1","metallophosphoesterase domain containing 2","claudin 18","zinc finger and BTB domain containing 11","ataxin 3","golgin A5","fibroblast growth factor receptor 2","leucine rich repeat containing 40","isochorismatase domain containing 1","EMAP like 1","tRNA methyltransferase 11 homolog","THUMP domain containing 1","Myb/SANT DNA binding domain containing 3","autophagy related 2B","ADP ribosylation factor guanine nucleotide exchange factor 1","acyl-CoA synthetase medium chain family member 2B","zinc finger and AT-hook domain containing","mitochondrial fission regulator 1","stromal antigen 3","ferrochelatase","myosin IXA","DEAD-box helicase 3 Y-linked","phosphofructokinase, platelet","isopentenyl-diphosphate delta isomerase 1","SP100 nuclear antigen","Kruppel like factor 6","phospholipid phosphatase 1","neogenin 1","translocation associated membrane protein 1","phosphorylase kinase regulatory subunit alpha 1","TNF receptor superfamily member 1A","calcium voltage-gated channel auxiliary subunit beta 1","ecotropic viral integration site 5","stomatin like 1","pyruvate kinase M1/2","DExH-box helicase 29","deoxynucleotidyltransferase terminal interacting protein 2","methyltransferase like 22","tumor protein p53 binding protein 1","trophinin","ribosomal RNA processing 15 homolog","ras homolog family member A","DEAH-box helicase 8","protein kinase C zeta","zinc finger protein Y-linked","isoleucyl-tRNA synthetase 2, mitochondrial","neuron navigator 3","isocitrate dehydrogenase (NAD(+)) 3 non-catalytic subunit gamma","rogdi atypical leucine zipper","PDZ domain containing 4","Rho associated coiled-coil containing protein kinase 1","core-binding factor subunit beta","pyruvate dehydrogenase kinase 3","hyaluronidase 2","histone deacetylase 4","Ras association domain family member 1","fibroblast growth factor receptor 3","interferon induced protein 35","HEAT repeat containing 6","Coenzyme A synthase","pleckstrin homology, MyTH4 and FERM domain containing H3","myocyte enhancer factor 2A","OTU deubiquitinase 5","transcription factor binding to IGHM enhancer 3","TBC1 domain family member 25","acyl-CoA synthetase long chain family member 4","inositol polyphosphate-5-phosphatase A","G-patch domain and KOW motifs","GRIP1 associated protein 1","FtsJ RNA 2'-O-methyltransferase 1","proline rich 11","receptor accessory protein 1","ATPase phospholipid transporting 11A","RNA polymerase I subunit A","lysosomal protein transmembrane 4 alpha","tetratricopeptide repeat domain 7A","inositol hexakisphosphate kinase 2","S1 RNA binding domain 1","kinesin family member 2A","RAS guanyl releasing protein 2","proteasome activator subunit 4","intraflagellar transport 80","sirtuin 2","endoplasmic reticulum lectin 1","protein phosphatase 2 regulatory subunit B'beta","glycogen phosphorylase, muscle associated","paired like homeodomain 1","microtubule associated serine/threonine kinase family member 4","adhesion G protein-coupled receptor F5","sidekick cell adhesion molecule 2","nucleoporin 133","nuclear casein kinase and cyclin dependent kinase substrate 1","VPS35 retromer complex component","DnaJ heat shock protein family (Hsp40) member A2","BCL3 transcription coactivator","potassium voltage-gated channel subfamily A regulatory beta subunit 2","ATP binding cassette subfamily C member 9","C-type lectin domain family 2 member D","FUN14 domain containing 1","monoamine oxidase B","RAR related orphan receptor A","dopamine receptor D4","transforming growth factor beta receptor 3","phospholipase A2 group X","hes family bHLH transcription factor 2","ATPase Na+/K+ transporting subunit beta 3","NEDD4 E3 ubiquitin protein ligase","phosphatidylinositol glycan anchor biosynthesis class B","mitogen-activated protein kinase 6","G protein subunit beta 5","RAB27A, member RAS oncogene family","haloacid dehalogenase like hydrolase domain containing 5","ubiquitin recognition factor in ER associated degradation 1","LDL receptor related protein 6","guanylate cyclase 2C","PHD and ring finger domains 1","elongator complex protein 1","nucleobindin 2","profilin 2","protein tyrosine phosphatase non-receptor type 3","spectrin beta, erythrocytic","dual adaptor of phosphotyrosine and 3-phosphoinositides 1","fibroblast growth factor 10","solute carrier family 44 member 1","transmembrane protein 260","SMG6 nonsense mediated mRNA decay factor","exocyst complex component 5","clathrin heavy chain like 1","follistatin like 3","DiGeorge syndrome critical region gene 2","ring finger protein 126","MAX network transcriptional repressor","ZXD family zinc finger C","jumonji domain containing 6, arginine demethylase and lysine hydroxylase","DNA polymerase beta","ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 1","WD repeat domain, phosphoinositide interacting 1","FERM and PDZ domain containing 1","glucosylceramidase beta 2","N-deacetylase and N-sulfotransferase 1","asparagine synthetase (glutamine-hydrolyzing)","adaptor related protein complex 3 subunit mu 2","cyclic nucleotide gated channel subunit beta 1","ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 2","choline O-acetyltransferase","poly(A) binding protein cytoplasmic 1","testis associated actin remodelling kinase 2","cilia and flagella associated protein 20","casein kinase 2 alpha 2","protein tyrosine phosphatase non-receptor type 21","eukaryotic translation initiation factor 2B subunit gamma","calcium/calmodulin dependent protein kinase II alpha","treacle ribosome biogenesis factor 1","cell division cycle 42","oxysterol binding protein like 3","solute carrier family 12 member 3","RAD18 E3 ubiquitin protein ligase","ATPase plasma membrane Ca2+ transporting 1","transient receptor potential cation channel subfamily M member 5","NCK adaptor protein 2","mitogen-activated protein kinase kinase kinase kinase 4","alpha-1,3-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase A","ribosomal protein L31","WD repeat domain 1","sorting nexin 13","Rho GTPase activating protein 10","ribosomal protein S6 kinase A2","inhibitor of growth family member 3","vasohibin 1","LIM and cysteine rich domains 1","BUD23 rRNA methyltransferase and ribosome maturation factor","SEL1L adaptor subunit of ERAD E3 ubiquitin ligase","thyroid hormone receptor interactor 13","ATPase H+ transporting accessory protein 1","transcription factor 3","tribbles pseudokinase 2","DAZ associated protein 1","methyl-CpG binding domain protein 3","helicase like transcription factor","family with sequence similarity 50 member A","family with sequence similarity 3 member A","cleavage and polyadenylation specific factor 1","myosin IIIB","cytochrome b reductase 1","cadherin 19","programmed cell death 2","solute carrier family 6 member 15","retinol dehydrogenase 11","protein kinase cAMP-activated catalytic subunit alpha","adhesion G protein-coupled receptor L1","secreted phosphoprotein 2","actinin alpha 1","zinc finger FYVE-type containing 26","ribosomal protein S6 kinase A6","epsin 2","protein tyrosine phosphatase non-receptor type 18","LIM zinc finger domain containing 2","acid sensing ion channel subunit family member 4","striated muscle enriched protein kinase","ligand of numb-protein X 1","aldehyde dehydrogenase 3 family member A2","transferrin receptor","sterol regulatory element binding transcription factor 1","transient receptor potential cation channel subfamily C member 5","AF4/FMR2 family member 4","ubiquitin conjugating enzyme E2 D1","membrane palmitoylated protein 5","Rho related BTB domain containing 1","structural maintenance of chromosomes 1A","hydroxysteroid 17-beta dehydrogenase 10","microtubule affinity regulating kinase 2","hyaluronan mediated motility receptor","checkpoint with forkhead and ring finger domains","thyrotropin releasing hormone degrading enzyme","prolyl 4-hydroxylase subunit alpha 2","Fc fragment of IgG receptor IIb","nuclear factor of activated T cells 3","tRNA nucleotidyl transferase 1","acyl-CoA dehydrogenase very long chain","serine/threonine kinase 10","F-box and WD repeat domain containing 11","ArfGAP with coiled-coil, ankyrin repeat and PH domains 1","collapsin response mediator protein 1","EvC ciliary complex subunit 1","derlin 2","SID1 transmembrane family member 1","nudE neurodevelopment protein 1","murine retrovirus integration site 1 homolog","transmembrane protein 38A","adaptor related protein complex 1 subunit mu 1","PVR cell adhesion molecule","X-ray repair cross complementing 1","scavenger receptor class B member 1","cytochrome P450 family 2 subfamily W member 1","minichromosome maintenance complex component 2","pannexin 2","selenoprotein O","tumor protein p63","alpha kinase 1","LLGL scribble cell polarity complex component 2","phosphodiesterase 8A","chloride voltage-gated channel 4","notchless homolog 1","succinate dehydrogenase complex flavoprotein subunit A","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily e, member 1","gasdermin B","lysine demethylase 5A","protein phosphatase 2 regulatory subunit B''alpha","fermitin family member 2","ATP binding cassette subfamily B member 11","dehydrogenase/reductase 9","prostaglandin-endoperoxide synthase 2","insulin like growth factor 2 mRNA binding protein 2","mitogen-activated protein kinase kinase kinase 13","ST6 beta-galactoside alpha-2,6-sialyltransferase 1","T-box transcription factor 21","FRY microtubule binding protein","phosphatidylinositol binding clathrin assembly protein","N-ethylmaleimide sensitive factor, vesicle fusing ATPase","GLI family zinc finger 2","cytoplasmic linker associated protein 1","mitochondrial ribosomal protein S34","notch receptor 3","chloride nucleotide-sensitive channel 1A","protein phosphatase 2 regulatory subunit Bgamma","embryonic ectoderm development","cadherin related family member 2","tumor susceptibility 101","nuclear cap binding subunit 3","ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 3","carbonic anhydrase 12","monoglyceride lipase","netrin 4","BCS1 homolog, ubiquinol-cytochrome c reductase complex chaperone","NUAK family kinase 1","dipeptidyl peptidase 8","solute carrier family 24 member 1","zinc finger protein 532","scavenger receptor class F member 1","lectin, mannose binding 1","3-hydroxyacyl-CoA dehydratase 3","interaction protein for cytohesin exchange factors 1","zinc finger ZZ-type and EF-hand domain containing 1","enolase 1","solute carrier family 12 member 1","myeloid derived growth factor","anoctamin 8","tubulin epsilon 1","Rho guanine nucleotide exchange factor 10 like","TXK tyrosine kinase","tachykinin receptor 2","actin related protein 6","TIMELESS interacting protein","sorcin","eukaryotic translation initiation factor 4 gamma 3","nucleoporin 37","semaphorin 3A","G2 and S-phase expressed 1","semaphorin 3C","tetratricopeptide repeat domain 38","acetyl-CoA acetyltransferase 1","GRAM domain containing 4","cadherin EGF LAG seven-pass G-type receptor 1","zinc finger protein 638","solute carrier family 25 member 40","translocase of inner mitochondrial membrane 21","adducin 2","RAS protein activator like 2","VPS9 domain containing 1","zinc finger protein 37A","microtubule affinity regulating kinase 3","solute carrier family 25 member 3","fibronectin type III domain containing 3B","FOS like 2, AP-1 transcription factor subunit","calcium voltage-gated channel auxiliary subunit gamma 5","calcium voltage-gated channel auxiliary subunit gamma 4","FRY like transcription coactivator","transmembrane protein 131","fascin actin-bundling protein 1","actin beta","molybdenum cofactor sulfurase","phospholipase D1","ATPase H+/K+ transporting non-gastric alpha2 subunit","WD repeat domain 62","discs large MAGUK scaffold protein 1","RAB7A, member RAS oncogene family","B cell receptor associated protein 29","SEC31 homolog B, COPII coat complex component","spliceosome associated factor 3, U4/U6 recycling protein","Rho GTPase activating protein 15","tubulin alpha 3d","exosome component 7","kinesin associated protein 3","makorin ring finger protein 2","minichromosome maintenance complex component 6","RNA exonuclease 2","RNA binding motif protein 7","RNA binding motif single stranded interacting protein 2","bromodomain adjacent to zinc finger domain 2A","protein tyrosine phosphatase non-receptor type 23","mutL homolog 1","uracil DNA glycosylase","flavin containing dimethylaniline monoxygenase 4","kelch like family member 20","regulator of G protein signaling 11","solute carrier family 46 member 1","plexin A2","sperm associated antigen 5","ankyrin repeat domain 13A","tumor protein D52","acetyl-CoA carboxylase beta","TNF receptor associated factor 4","phosphoprotein membrane anchor with glycosphingolipid microdomains 1","G-patch domain containing 1","intercellular adhesion molecule 3","5'-nucleotidase, cytosolic II","melanoma cell adhesion molecule","glypican 4","muscleblind like splicing regulator 3","calmodulin regulated spectrin associated protein family member 3","RAP1 GTPase activating protein","XPA binding protein 2","Rho guanine nucleotide exchange factor 1","syntaxin binding protein 2","mitogen-activated protein kinase kinase 7","diacylglycerol kinase delta","cortactin binding protein 2","retinoic acid receptor beta","DNA topoisomerase II beta","transmembrane 9 superfamily member 3","nuclear factor kappa B subunit 2","ubiquitin conjugating enzyme E2 T","protein phosphatase 1 regulatory subunit 12B","DnaJ heat shock protein family (Hsp40) member C10","general transcription factor IIIC subunit 1","interleukin 4 receptor","ubiquitin specific peptidase 33","p21 (RAC1) activated kinase 3","doublecortin","small nuclear ribonucleoprotein polypeptide A","sperm associated antigen 6","exosome component 5","dynein cytoplasmic 1 intermediate chain 2","amyloid beta precursor protein binding family B member 1 interacting protein","leucine rich repeats and calponin homology domain containing 4","family with sequence similarity 76 member B","sirtuin 6","tyrosinase","DNA polymerase delta 3, accessory subunit","capping actin protein of muscle Z-line subunit beta","G protein-coupled receptor 137B","N-acetylated alpha-linked acidic dipeptidase 2","jade family PHD finger 1","solute carrier family 25 member 43","ubiquitin conjugating enzyme E2 A","fibroblast growth factor receptor 1","FKBP prolyl isomerase 6","structural maintenance of chromosomes 1B","fibulin 1","integrin subunit alpha 8","cystatin F","microtubule associated protein 2","protein inhibitor of activated STAT 2","amphiphysin","A-Raf proto-oncogene, serine/threonine kinase","methylcrotonoyl-CoA carboxylase 1","lysosomal associated membrane protein 3","fibroblast activation protein alpha","nebulette","alkaline ceramidase 3","ubiquitin conjugating enzyme E2 K","phosphatidylinositol 3-kinase catalytic subunit type 3","NEDD4 binding protein 2","TP53 induced glycolysis regulatory phosphatase","TUB like protein 3","synaptojanin 2","adenylate cyclase 2","protein phosphatase 2 regulatory subunit B'gamma","RNA binding fox-1 homolog 1","G protein subunit beta 1","endothelin 1","MLLT10 histone lysine methyltransferase DOT1L cofactor","zinc finger CW-type and PWWP domain containing 1","P2Y receptor family member 10","integral membrane protein 2A","nardilysin convertase","voltage dependent anion channel 3","pericentriolar material 1","trinucleotide repeat containing adaptor 6C","CBFA2/RUNX1 partner transcriptional co-repressor 2","itchy E3 ubiquitin protein ligase","polycystin 2 like 2, transient receptor potential cation channel","tumor protein p53 inducible nuclear protein 2","stromal cell derived factor 4","myosin heavy chain 7B","BPI fold containing family B member 2","tumor protein p73","toll interacting protein","ubiquitin conjugating enzyme E2 D4 (putative)","clusterin like 1","RUNX1 partner transcriptional co-repressor 1","cadherin 17","THO complex 1","FKBP prolyl isomerase 7","oxysterol binding protein like 6","solute carrier family 1 member 3","X-ray repair cross complementing 5","latexin","SP140 nuclear body protein","MAPK interacting serine/threonine kinase 1","tensin 1","RNA exonuclease 1 homolog","secretion associated Ras related GTPase 1A","cell division cycle 14A","Rap guanine nucleotide exchange factor 3","CEA cell adhesion molecule 1","SUMO specific peptidase 1","capicua transcriptional repressor","lipase E, hormone sensitive type","farnesyl-diphosphate farnesyltransferase 1","platelet activating factor acetylhydrolase 1b catalytic subunit 3","oligophrenin 1","kinesin family member 22","capping protein regulator and myosin 1 linker 1","phosphoglucomutase 1","DEAD-box helicase 1","dynamin 2","erythrocyte membrane protein band 4.1 like 2","regulating synaptic membrane exocytosis 1","syntaxin 7","RAB, member of RAS oncogene family like 2B","kelch like ECH associated protein 1","DEAD-box helicase 43","protein tyrosine phosphatase receptor type H","solute carrier family 35 member C2","crystallin beta-gamma domain containing 3","secretin receptor","regulatory factor X3","replication timing regulatory factor 1","RAB21, member RAS oncogene family","solute carrier family 4 member 4","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 2","sestrin 1","midline 2","PIH1 domain containing 3","Snf2 related CREBBP activator protein","pumilio RNA binding family member 3","cholinergic receptor nicotinic alpha 3 subunit","potassium calcium-activated channel subfamily N member 2","CCR4-NOT transcription complex subunit 4","presenilin 1","coproporphyrinogen oxidase","claudin domain containing 1","MOK protein kinase","heat shock protein 90 alpha family class A member 1","RB transcriptional corepressor like 1","DLG associated protein 4","immunoglobulin superfamily member 9B","NDC80 kinetochore complex component","adaptor related protein complex 4 subunit epsilon 1","round spermatid basic protein 1","membrane associated guanylate kinase, WW and PDZ domain containing 3","C-X-C motif chemokine ligand 2","collagen type IV alpha 4 chain","transcription factor 7","osteoclastogenesis associated transmembrane protein 1","cadherin 7","interphotoreceptor matrix proteoglycan 2","PEST proteolytic signal containing nuclear protein","exonuclease 3'-5' domain containing 2","arginase 2","myocyte enhancer factor 2C","protein tyrosine phosphatase receptor type C","plakophilin 1","ubiquitin like modifier activating enzyme 5","serine/threonine kinase 17b","cell division cycle 14B","zinc finger protein 510","LDL receptor related protein 2","zinc finger protein 506","jumonji domain containing 4","dual specificity phosphatase 12","acetoacetyl-CoA synthetase","DAP3 binding cell death enhancer 1","calcium dependent secretion activator 2","protocadherin beta 4","heat shock protein family B (small) member 11","PH domain and leucine rich repeat protein phosphatase 1","ATPase phospholipid transporting 8B1","interleukin 12 receptor subunit beta 2","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 3","WD repeat domain 70","FYN binding protein 1","membrane palmitoylated protein 4","STE20 related adaptor beta","basic leucine zipper and W2 domains 1","C1q and TNF related 3","malic enzyme 2","chromosome 5 open reading frame 22","cyclin T2","family with sequence similarity 135 member A","collagen type XIX alpha 1 chain","erythrocyte membrane protein band 4.1 like 3","cordon-bleu WH2 repeat protein like 1","discs large MAGUK scaffold protein 3","potassium two pore domain channel subfamily K member 2","SERTA domain containing 4","TNF receptor associated factor 5","mitochondrial ribosomal protein L22","gem nuclear organelle associated protein 5","nuclear factor, erythroid 2 like 1","semaphorin 5B","glycogen synthase kinase 3 beta","integrin subunit beta 5","ELKS/RAB6-interacting/CAST family member 1","exportin 1","ring finger protein 13","transient receptor potential cation channel subfamily M member 3","partner and localizer of BRCA2","DOP1 leucine zipper like protein A","LYR motif containing 2","branched chain keto acid dehydrogenase E1 subunit beta","lysine acetyltransferase 6A","terminal uridylyl transferase 7","unc-51 like autophagy activating kinase 2","grainyhead like transcription factor 2","transportin 1","procollagen-lysine,2-oxoglutarate 5-dioxygenase 1","purinergic receptor P2X 5","integrin subunit alpha E","DIS3 homolog, exosome endoribonuclease and 3'-5' exoribonuclease","progesterone immunomodulatory binding factor 1","tudor domain containing 3","nuclear FMR1 interacting protein 1","PDS5 cohesin associated factor B","3-oxoacid CoA-transferase 1","Ras related GTP binding B","CYLD lysine 63 deubiquitinase","solute carrier family 27 member 5","zinc finger protein 324","zinc finger protein 671","zinc finger protein 416","zinc finger protein 586","zinc finger protein 446","zinc finger protein 264","ribosomal protein S5","FAT atypical cadherin 1","YTH domain containing 1","charged multivesicular body protein 2B","small ArfGAP2","peptidylprolyl isomerase E","zinc metallopeptidase STE24","StAR related lipid transfer domain containing 7","nitric oxide associated 1","RE1 silencing transcription factor","histidine ammonia-lyase","slingshot protein phosphatase 1","glutathione S-transferase pi 1","amyloid beta precursor like protein 2","family with sequence similarity 234 member B","solute carrier organic anion transporter family member 1A2","WW domain binding protein 11","eukaryotic translation initiation factor 3 subunit I","collagen type XVI alpha 1 chain","taxilin alpha","apolipoprotein B","nuclear receptor coactivator 1","ATP/GTP binding protein like 5","EFR3 homolog B","kinesin family member 3C","RAB10, member RAS oncogene family","glucokinase regulator","hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha","microtubule associated protein RP/EB family member 3","carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase","CD59 molecule (CD59 blood group)","CD82 molecule","BCL6 corepressor like 1","ATRX chromatin remodeler","adenylate kinase 6","ficolin 1","myoneurin","MDS1 and EVI1 complex locus","secretory carrier membrane protein 1","prolyl endopeptidase","HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1","SEH1 like nucleoporin","WD repeat domain 47","WD repeat and FYVE domain containing 1","oviductal glycoprotein 1","solute carrier family 25 member 24","mitogen-activated protein kinase kinase kinase 4","paired immunoglobin like type 2 receptor alpha","immunoglobulin superfamily member 9","ATP binding cassette subfamily B member 1","zinc finger protein 213","aldo-keto reductase family 1 member B","copine 3","RRN3 homolog, RNA polymerase I transcription factor","cortactin","mitochondrial translational initiation factor 2","DDHD domain containing 2","tetratricopeptide repeat domain 39A","epidermal growth factor receptor pathway substrate 15","origin recognition complex subunit 1","microsomal glutathione S-transferase 2","calcium homeostasis endoplasmic reticulum protein","autophagy related 16 like 1","ubiquitin specific peptidase 40","protein O-linked mannose N-acetylglucosaminyltransferase 1 (beta 1,2-)","RAD54 like","microtubule associated serine/threonine kinase 2","DnaJ heat shock protein family (Hsp40) member A1","beta-1,4-galactosyltransferase 1","charged multivesicular body protein 5","nuclear transcription factor, X-box binding 1","aquaporin 6","DIMT1 rRNA methyltransferase and ribosome maturation factor","importin 11","folate hydrolase 1","eukaryotic translation initiation factor 2 alpha kinase 1","ependymin related 1","sorting nexin 10","selenophosphate synthetase 1","mitochondrial ribosomal protein L28","inositol-trisphosphate 3-kinase C","CEA cell adhesion molecule 6","FAT atypical cadherin 2","RNA binding motif protein 22","transmembrane p24 trafficking protein 2","endoplasmic reticulum oxidoreductase 1 beta","zinc finger AN1-type containing 6","hydroxysteroid 17-beta dehydrogenase 2","taxilin gamma","linker for activation of T cells family member 2","HECT, UBA and WWE domain containing E3 ubiquitin protein ligase 1","zw10 kinetochore protein","ALG9 alpha-1,2-mannosyltransferase","NADPH oxidase 4","acyl-CoA oxidase 3, pristanoyl","myotubularin related protein 2","protein phosphatase 1 regulatory subunit 15A","thyroid hormone receptor interactor 6","acetylcholinesterase (Cartwright blood group)","ferritin light chain","serrate, RNA effector molecule","BCL2 associated X, apoptosis regulator","nemo like kinase","phosphatidylinositol glycan anchor biosynthesis class S","ADAM metallopeptidase with thrombospondin type 1 motif 2","transmembrane serine protease 11E","ataxin 7 like 3","phosphatidylglycerophosphate synthase 1","proteasome 26S subunit, ATPase 5","ubiquitin interaction motif containing 1","cholesteryl ester transfer protein","metallothionein 3","lysophosphatidylcholine acyltransferase 2","G protein subunit alpha o1","2-oxoglutarate and iron dependent oxygenase domain containing 1","SH3 domain binding protein 2","NOP14 nucleolar protein","adducin 1","L-2-hydroxyglutarate dehydrogenase","thioredoxin domain containing 16","RNA transcription, translation and transport factor","nidogen 2","germ cell-less 1, spermatogenesis associated","splicing factor 3b subunit 2","kelch like family member 42","GNAS complex locus","dynamin 1 like","phosphatase and actin regulator 3","ERGIC and golgi 2","transcription factor AP-2 gamma","aurora kinase A","Cas scaffold protein family member 4","pirin","adipogenesis associated Mth938 domain containing","regulatory factor X2","methyltransferase like 2A","sulfotransferase family 2B member 1","ALG6 alpha-1,3-glucosyltransferase","CCR4-NOT transcription complex subunit 3","glycoprotein VI platelet","protein tyrosine phosphatase non-receptor type 4","DEAD-box helicase 18","KH-type splicing regulatory protein","G protein subunit alpha 11","ArfGAP with SH3 domain, ankyrin repeat and PH domain 3","ER degradation enhancing alpha-mannosidase like protein 2","DNA methyltransferase 3 beta","RRAD and GEM like GTPase 1","TPX2 microtubule nucleation factor","p53 and DNA damage regulated 1","erythrocyte membrane protein band 4.1 like 1","solute carrier family 15 member 1","dedicator of cytokinesis 9","ankyrin repeat domain 10","TDP-glucose 4,6-dehydratase","dedicator of cytokinesis 3","chromosome 3 open reading frame 18","coenzyme Q9","transmembrane protein 40","kinesin family member 9","Rho GTPase activating protein 28","cardiolipin synthase 1","protein phosphatase 1 regulatory subunit 13B","attractin","spermine oxidase","sialic acid binding Ig like lectin 1","FKBP prolyl isomerase 1A","NSFL1 cofactor","solute carrier family 4 member 11","chromosome 20 open reading frame 194","zinc finger protein 343","EBF family member 4","carboxypeptidase X, M14 family member 1","mitochondrial antiviral signaling protein","leucine zipper tumor suppressor family member 3","5'-3' exoribonuclease 2","kizuna centrosomal protein","dynein light chain LC8-type 1","tescalcin","sorting nexin 5","ribosomal protein L6","signal regulatory protein gamma","MAPK activated protein kinase 5","purinergic receptor P2X 7","ESF1 nucleolar pre-rRNA processing protein homolog","RB binding protein 9, serine hydrolase","anaphase promoting complex subunit 5","solute carrier family 23 member 2","solute carrier family 8 member B1","transmembrane protein 230","double zinc ribbon and ankyrin repeat domains 1","lysine demethylase 2B","cilia and flagella associated protein 61","taspase 1","2'-5'-oligoadenylate synthetase 1","GCN1 activator of EIF2AK4","ribosomal protein lateral stalk subunit P0","paxillin","rabphilin 3A","kinesin family member 16B","tRNA methyltransferase 6","chromogranin B","phosphatidylethanolamine binding protein 1","BRCA1 associated protein","endoplasmic reticulum protein 29","nitric oxide synthase 1","FUS RNA binding protein","immunoglobulin binding protein 1","FXYD domain containing ion transport regulator 5","zinc finger protein 302","GRAM domain containing 1A","FXYD domain containing ion transport regulator 3","cell death inducing p53 target 1","CKLF like MARVEL transmembrane domain containing 1","potassium voltage-gated channel subfamily H member 4","glucosidase II alpha subunit","GEM interacting protein","RNA binding motif protein 41","baculoviral IAP repeat containing 5","lymphocyte activating 3","myeloid leukemia factor 2","OTU deubiquitinase, ubiquitin aldehyde binding 2","DEAD-box helicase 24","zinc finger and BTB domain containing 25","NECAP endocytosis associated 1","Rho GTPase activating protein 4","ankyrin repeat domain 24","DEAH-box helicase 32 (putative)","REST corepressor 1","G-patch domain containing 2 like","latent transforming growth factor beta binding protein 4","biliverdin reductase B","solute carrier family 9 member A1","serine palmitoyltransferase long chain base subunit 1","poly(A) polymerase alpha","cyclin K","poly(rC) binding protein 4","regulator of G protein signaling 1","yippee like 3","mitochondrial ribosomal protein S33","NADH:ubiquinone oxidoreductase subunit B2","nuclear distribution C, dynein complex regulator","macrophage erythroblast attacher","intercellular adhesion molecule 1","striatin 4","interleukin 1 receptor associated kinase 3","lysozyme","mitochondrial E3 ubiquitin protein ligase 1","transcription factor AP-4","programmed cell death 7","SPG21 abhydrolase domain containing, maspardin","DnaJ heat shock protein family (Hsp40) member B11","prolyl 3-hydroxylase 2","chordin","fms related tyrosine kinase 3 ligand","RAB11 family interacting protein 3","N-acetylglucosamine-1-phosphate transferase subunit gamma","zinc finger protein 268","golgin A3","poly(A) binding protein cytoplasmic 4","CD209 molecule","ceramide synthase 4","mucolipin 1","ubiquitin specific peptidase 48","ephrin B1","pyruvate dehydrogenase phosphatase regulatory subunit","alanyl-tRNA synthetase 1","golgi glycoprotein 1","kinesin family member 4A","trinucleotide repeat containing adaptor 6A","pleckstrin homology and RhoGEF domain containing G2","N-acetyltransferase 14 (putative)","phosphatidylinositol transfer protein membrane associated 2","exocyst complex component 1","RNA binding motif protein 27","oxysterol binding protein like 8","deltex E3 ubiquitin ligase 2","NLR family CARD domain containing 4","pseudouridine synthase 7","neuronal cell adhesion molecule","laminin subunit beta 1","solute carrier family 26 member 4","dihydrolipoamide dehydrogenase","WD repeat domain 7","thioredoxin like 1","interleukin 5 receptor subunit alpha","ATP binding cassette subfamily C member 6","CKLF like MARVEL transmembrane domain containing 6","integrin subunit alpha 6","Rap guanine nucleotide exchange factor 4","mitogen-activated protein kinase kinase kinase 20","fumarate hydratase","SEL1L family member 3","transferrin","CDV3 homolog","myosin XVA","alkB homolog 5, RNA demethylase","NLR family pyrin domain containing 1","PITPNM family member 3","sperm associated antigen 7","origin recognition complex subunit 6","zinc finger homeobox 4","solute carrier family 17 member 6","zinc finger C3HC-type containing 1","estrogen receptor 1","regulator of G protein signaling 17","angiopoietin 2","transmembrane protein 101","CD200 molecule","coiled-coil domain containing 80","proteasome activator subunit 1","protein phosphatase 2 regulatory subunit B''gamma","HAUS augmin like complex subunit 4","solute carrier family 7 member 8","O-sialoglycoprotein endopeptidase","solute carrier family 22 member 17","ring finger protein 31","sec1 family domain containing 1","G2/M-phase specific E3 ubiquitin protein ligase","HECT domain E3 ubiquitin protein ligase 1","heterogeneous nuclear ribonucleoprotein C","RPGR interacting protein 1","SPT16 homolog, facilitates chromatin remodeling subunit","TOX high mobility group box family member 4","gem nuclear organelle associated protein 2","transglutaminase 1","TERF1 interacting nuclear factor 2","semaphorin 6A","transient receptor potential cation channel subfamily M member 7","TYRO3 protein tyrosine kinase","WD repeat domain 76","calpain 3","synaptosome associated protein 23","phosphoglycerate dehydrogenase","collagen type IX alpha 3 chain","ezrin","myosin light chain 6","argonaute RISC component 1","tektin 2","claspin","ring finger and FYVE like domain containing E3 ubiquitin protein ligase","unc-13 homolog D","major facilitator superfamily domain containing 11","dihydropyrimidinase like 2","transforming growth factor beta 2","G-patch domain containing 2","nucleoporin 50","catechol-O-methyltransferase","adenosine deaminase 2","vanin 3","ethylmalonyl-CoA decarboxylase 1","LRR binding FLII interacting protein 2","SEC22 homolog C, vesicle trafficking protein","xylulokinase","histone deacetylase 6","olfactory receptor family 1 subfamily I member 1","gamma-aminobutyric acid type A receptor pi subunit","cell division cycle 6","uracil phosphoribosyltransferase homolog","cell division cycle 23","aladin WD repeat nucleoporin","chromobox 5","flavin containing dimethylaniline monoxygenase 2","SUN domain containing ossification factor","mutS homolog 2","mitogen-activated protein kinase kinase kinase 1","deoxyhypusine synthase","hook microtubule tethering protein 2","archain 1","erythrocyte membrane protein band 4.1 like 4B","transmembrane protein 38B","proteasome 26S subunit, non-ATPase 5","prostaglandin-endoperoxide synthase 1","nucleoporin 188","carnitine O-acetyltransferase","SH2 domain containing 3C","N-acetylneuraminate synthase","TBC1 domain family member 2","whirlin","phosphodiesterase 6C","CWF19 like cell cycle control factor 1","semaphorin 4G","B-TFIID TATA-box binding protein associated factor 1","IKAROS family zinc finger 5","B cell linker","tolloid like 2","cytochrome P450 family 26 subfamily A member 1","tudor domain containing 1","sorbin and SH3 domain containing 1","BMP and activin membrane bound inhibitor","WW domain containing adaptor with coiled-coil","cAMP responsive element modulator","nucleotide binding protein 2","HIVEP zinc finger 1","triggering receptor expressed on myeloid cells 2","cysteine rich secretory protein 3","FKBP prolyl isomerase 5","SRSF protein kinase 1","bromodomain and PHD finger containing 3","mitochondrial ribosomal protein S18A","transmembrane protein 14A","EF-hand domain containing 1","heat shock protein 90 alpha family class B member 1","cell division cycle 5 like","inositol 1,4,5-trisphosphate receptor type 3","zinc finger protein 184","desmoplakin","sirtuin 1","heterogeneous nuclear ribonucleoprotein H3","intraflagellar transport 74","Janus kinase 2","interleukin 12 receptor subunit beta 1","ABL proto-oncogene 1, non-receptor tyrosine kinase","acyl-CoA thioesterase 7","SH3 domain containing GRB2 like, endophilin B1","cell division cycle 7","synapse defective Rho GTPase homolog 2","proprotein convertase subtilisin/kexin type 5","stearoyl-CoA desaturase","transmembrane p24 trafficking protein 1","actin binding LIM protein 1","endoplasmic reticulum metallopeptidase 1","RAB18, member RAS oncogene family","neuropilin 1","palmdelphin","tetraspanin 15","macroH2A.2 histone","WASH complex subunit 2A","microtubule associated serine/threonine kinase 3","myeloid zinc finger 1","occludin/ELL domain containing 1","myosin IXB","potassium two pore domain channel subfamily K member 6","cation channel sperm associated auxiliary subunit gamma","proteasome 26S subunit, non-ATPase 8","F-box and leucine rich repeat protein 19","syntaxin 1B","hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 7","SET domain containing 1A, histone lysine methyltransferase","BAF chromatin remodeling complex subunit BCL7C","ephrin A2","cold inducible RNA binding protein","ATP synthase F1 subunit delta","CACN subunit beta associated regulatory protein","protocadherin 11 Y-linked","protein kinase Y-linked (pseudogene)","insulin like growth factor binding protein acid labile subunit","heterogeneous nuclear ribonucleoprotein M","membrane associated ring-CH-type finger 2","NADH:ubiquinone oxidoreductase subunit B7","trans-2,3-enoyl-CoA reductase","translocase of inner mitochondrial membrane 13","cell division cycle 34","methylthioadenosine phosphorylase","mitotic spindle positioning","centrosomal protein 170B","RNA polymerase II subunit E","RNA polymerase mitochondrial","cadherin related family member 5","IZUMO family member 4","Ras association domain family member 7","growth arrest and DNA damage inducible beta","paralemmin","mucosal vascular addressin cell adhesion molecule 1","MAPK interacting serine/threonine kinase 2","ARVCF delta catenin family member","tRNA methyltransferase 2 homolog A","RAN binding protein 1","zinc finger DHHC-type containing 8","kelch like family member 22","mediator complex subunit 15","serpin family D member 1","synaptosome associated protein 29","CRK like proto-oncogene, adaptor protein","leucine zipper like transcription regulator 1","CECR2 histone acetyl-lysine reader","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1","purinergic receptor P2X 6","derlin 3","solute carrier family 7 member 4","BCL2 like 13","D-dopachrome tautomerase like","D-dopachrome tautomerase","oncostatin M","calcineurin binding protein 1","TBC1 domain family member 10A","sushi domain containing 2","splicing factor 3a subunit 1","gamma-glutamyltransferase 5","ring finger protein 215","SEC14 like lipid binding 2","SEC14 like lipid binding 3","sperm antigen with calponin homology and coiled-coil domains 1 like","peptidylprolyl isomerase like 2","beta-ureidopropionase 1","yippee like 1","small nuclear ribonucleoprotein D3 polypeptide","pescadillo ribosomal biogenesis factor 1","mitogen-activated protein kinase 1","gamma-glutamyltransferase 1","proline dehydrogenase 1","protein phosphatase, Mg2+/Mn2+ dependent 1F","solute carrier family 35 member E4","DNA topoisomerase III beta","crystallin beta B3","cytohesin 4","ess-2 splicing factor homolog","MFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase","caspase recruitment domain family member 10","LDL receptor related protein 5 like","solute carrier family 25 member 1","G protein-coupled receptor kinase 3","galectin 2","golgi associated, gamma adaptin ear containing, ARF binding protein 1","histone cell cycle regulator","SH3 domain binding protein 1","galectin 1","HPS4 biogenesis of lysosomal organelles complex 3 subunit 2","phosphoinositide-3-kinase interacting protein 1","SRR1 domain containing","POZ/BTB and AT hook containing zinc finger 1","TRIO and F-actin binding protein","tuftelin interacting protein 11","glycine C-acetyltransferase","gamma-glutamyltransferase light chain 2","ankyrin repeat domain 54","eukaryotic translation initiation factor 3 subunit L","small nuclear ribonucleoprotein 13","MICAL like 1","RNA polymerase II subunit F","SRY-box transcription factor 10","coiled-coil domain containing 134","DEP domain containing 5, GATOR1 subcomplex subunit","protein interacting with PRKCA 1","tetratricopeptide repeat domain 28","solute carrier family 16 member 8","centromere protein M","septin 3","solute carrier family 5 member 1","KDEL endoplasmic reticulum protein retention receptor 3","cytochrome P450 family 2 subfamily D member 6","DEAD-box helicase 17","DNA meiotic recombinase 1","transcription factor 20","HscB mitochondrial iron-sulfur cluster cochaperone","chibby family member 1, beta catenin antagonist","translocase of outer mitochondrial membrane 22","radial spoke head 14 homolog","X-box binding protein 1","RNA 2',3'-cyclic phosphate and 5'-OH ligase","Josephin domain containing 1","F-box protein 7","GTP binding protein 1","DNA polymerase delta interacting protein 3","RAB36, member RAS oncogene family","TIMP metallopeptidase inhibitor 3","protein phosphatase 6 regulatory subunit 2","SET binding factor 1","Sad1 and UNC84 domain containing 2","cytochrome b5 reductase 3","dynein axonemal light chain 4","myo-inositol oxygenase","lipase maturation factor 2","rhomboid domain containing 3","protein kinase C and casein kinase substrate in neurons 2","tubulin tyrosine ligase like 1","RAS like family 10 member A","adaptor related protein complex 1 subunit beta 1","HMG-box containing 4","target of myb1 membrane trafficking protein","neurofilament heavy","choline kinase beta","BCL2 interacting killer","heme oxygenase 1","malonyl-CoA-acyl carrier protein transacylase","THO complex 5","minichromosome maintenance complex component 5","arylsulfatase A","translocator protein","tubulin tyrosine ligase like 12","chromobox 7","platelet derived growth factor subunit B","ribosomal protein L3","zinc finger matrin-type 5","RNA binding fox-1 homolog 2","synaptogyrin 1","TGF-beta activated kinase 1 (MAP3K7) binding protein 1","activating signal cointegrator 1 complex subunit 2","myotubularin related protein 3","mitochondrial elongation factor 1","apolipoprotein L4","patatin like phospholipase domain containing 5","apolipoprotein L1","patatin like phospholipase domain containing 3","myosin heavy chain 9","SAMM50 sorting and assembly machinery component","thioredoxin 2","FAD dependent oxidoreductase domain containing 2","GRB2 related adaptor protein 2","eukaryotic translation initiation factor 3 subunit D","trinucleotide repeat containing adaptor 6B","small G protein signaling modulator 3","intraflagellar transport 27","KIAA0930","neutrophil cytosolic factor 4","colony stimulating factor 2 receptor beta common subunit","solute carrier family 25 member 17","family with sequence similarity 118 member A","potassium channel tetramerization domain containing 17","ST13 Hsp70 interacting protein","interleukin 2 receptor subunit beta","ring-box 1","E1A binding protein p300","L3MBTL histone methyl-lysine binding protein 2","chondroadherin like","Ran GTPase activating protein 1","zinc finger CCCH-type containing 7B","PHD finger protein 5A","aconitase 2","RNA polymerase III subunit H","tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase","phosphomannomutase 1","desumoylating isopeptidase 1","ceramide kinase","bromodomain containing 1","zinc finger BED-type containing 4","modulator of VRAC current 1","histone deacetylase 10","potassium two pore domain channel subfamily K member 10","abhydrolase domain containing 4","KH and NYN domain containing","FKBP prolyl isomerase 3","short chain dehydrogenase/reductase family 39U member 1","granzyme H","granzyme B","RNA binding motif protein 23","protein arginine methyltransferase 5","cochlin","adaptor related protein complex 4 subunit sigma 1","DNA polymerase epsilon 2, accessory subunit","valosin containing protein lysine methyltransferase","SOS Ras/Rho guanine nucleotide exchange factor 2","cyclin dependent kinase like 1","ninein","glycogen phosphorylase L","tripartite motif containing 9","proteasome 26S subunit, ATPase 6","glucosamine-phosphate N-acetyltransferase 1","DDHD domain containing 1","cornichon family AMPA receptor auxiliary protein 1","cell growth regulator with ring finger domain 1","ATPase H+ transporting V1 subunit D","coiled-coil domain containing 198","pleckstrin 2","phosphatidylinositol glycan anchor biosynthesis class H","proteasome 20S subunit alpha 3","vesicle transport through interaction with t-SNAREs 1B","translocase of inner mitochondrial membrane 9","glutathione S-transferase zeta 1","KIAA0586","transmembrane p24 trafficking protein family member 8","sterile alpha motif domain containing 15","activator of HSP90 ATPase activity 1","dishevelled associated activator of morphogenesis 1","serine palmitoyltransferase long chain base subunit 2","Ras and Rab interactor 3","legumain","alkB homolog 1, histone H2A dioxygenase","SNW domain containing 1","chromogranin A","inositol-tetrakisphosphate 1-kinase","dehydrogenase/reductase 7","protein phosphatase, Mg2+/Mn2+ dependent 1A","SIX homeobox 4","polypeptide N-acetylgalactosaminyltransferase 16","ankyrin repeat and SOCS box containing 2","centrosomal protein 128","ERH mRNA splicing and mitosis factor","hypoxia inducible factor 1 subunit alpha","sushi domain containing 6","serine and arginine rich splicing factor 5","solute carrier family 10 member 1","eukaryotic translation initiation factor 5","dicer 1, ribonuclease III","zinc finger FYVE-type containing 21","methylenetetrahydrofolate dehydrogenase, cyclohydrolase and formyltetrahydrofolate synthetase 1","T cell leukemia/lymphoma 1A","zinc finger CCCH-type containing 14","telomere maintenance 2","pecanex 1","bradykinin receptor B1","GSK3B interacting protein","VRK serine/threonine kinase 1","proteasome 26S subunit, ATPase 1","papilin, proteoglycan like sulfated glycoprotein","ribosomal protein S6 kinase A5","protein phosphatase 4 regulatory subunit 3A","chromosome 14 open reading frame 93","proteasome 20S subunit beta 5","YY1 transcription factor","apoptotic chromatin condensation inducer 1","cyclin B1 interacting protein 1","thyroid hormone receptor interactor 11","apurinic/apyrimidinic endodeoxyribonuclease 1","poly(A) binding protein nuclear 1","embryonal Fyn-associated substrate","Rho GTPase activating protein 5","cyclin dependent kinase 2 interacting protein","dehydrogenase/reductase 2","signal recognition particle 54","chromodomain helicase DNA binding protein 8","phosphoenolpyruvate carboxykinase 2, mitochondrial","protein only RNase P catalytic subunit","DDB1 and CUL4 associated factor 11","proteasome 20S subunit alpha 6","NFKB inhibitor alpha","ER membrane protein complex subunit 9","proteasome activator subunit 2","BRMS1 like transcriptional repressor","REC8 meiotic recombination protein","transmembrane 9 superfamily member 1","Sec23 homolog A, coat complex II component","guanosine monophosphate reductase 2","pinin, desmosome associated protein","Rab geranylgeranyltransferase subunit alpha","nuclear factor of activated T cells 4","phospholipid transfer protein","PDX1 C-terminal inhibiting factor 1","glutathione synthetase","matrix metallopeptidase 9","transient receptor potential cation channel subfamily C member 4 associated protein","glycogen phosphorylase B","abhydrolase domain containing 12","protein C receptor","GINS complex subunit 1","ninein like","CD40 molecule","ubiquinol-cytochrome c reductase complex assembly factor 1","zinc finger MYND-type containing 8","serum/glucocorticoid regulated kinase 2","intraflagellar transport 52","MYB proto-oncogene like 2","hepatocyte nuclear factor 4 alpha","NDRG family member 3","Src like adaptor 2","RAB5 interacting factor","nuclear factor of activated T cells 2","poly(A) binding protein cytoplasmic 1 like","serine/threonine kinase 4","spalt like transcription factor 4","activity dependent neuroprotector homeobox","prefoldin subunit 4","cleavage stimulation factor subunit 1","bone morphogenetic protein 7","ribonucleic acid export 1","TPD52 like 2","DnaJ heat shock protein family (Hsp40) member C5","negative elongation factor complex member C/D","cathepsin Z","pre-mRNA processing factor 6","tubulin beta 1 class VI","PRELI domain containing 3B","mitochondrial ribosome associated GTPase 2","proteasome 20S subunit alpha 7","solute carrier organic anion transporter family member 4A1","MRG domain binding protein","transcription factor like 5","death inducer-obliterator 1","GID complex subunit 8 homolog","solute carrier family 17 member 9","ADP ribosylation factor GTPase activating protein 1","collagen type XX alpha 1 chain","eukaryotic translation elongation factor 1 alpha 2","protein tyrosine kinase 6","glucocorticoid modulatory element binding protein 2","chromosome 20 open reading frame 27","sperm flagellar 1","cell division cycle 25B","ring finger protein 24","ADP ribosylation factor related protein 1","NADH:ubiquinone oxidoreductase complex assembly factor 5","tribbles pseudokinase 3","Ras association domain family member 2","casein kinase 2 alpha 1","solute carrier family 52 member 3","angiopoietin 4","CDP-diacylglycerol synthase 2","histocompatibility minor 13","syntaphilin","signal regulatory protein beta 1","SEC23 homolog B, coat complex II component","fermitin family member 1","phospholipase C beta 4","myosin light chain 9","HCK proto-oncogene, Src family tyrosine kinase","transmembrane 9 superfamily member 4","TBC/LysM-associated domain containing 2","crooked neck pre-mRNA splicing factor 1","protein O-fucosyltransferase 1","SAM and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1","kinesin family member 3B","maestro heat like repeat family member 8","NOP56 ribonucleoprotein","mannosidase beta like","isocitrate dehydrogenase (NAD(+)) 3 non-catalytic subunit beta","microtubule associated protein RP/EB family member 1","jagged canonical Notch ligand 1","CDK5 regulatory subunit associated protein 1","syntrophin alpha 1","TELO2 interacting protein 1","E2F transcription factor 1","regulation of nuclear pre-mRNA domain containing 1B","peroxisomal membrane protein 4","charged multivesicular body protein 4B","bactericidal permeability increasing protein","cystatin C","actin related protein 5","WAP four-disulfide core domain 2","adenosylhomocysteinase","protein phosphatase 1 regulatory subunit 16B","family with sequence similarity 83 member D","epididymal peptidase inhibitor","DEAH-box helicase 35","deoxynucleotidyltransferase terminal interacting protein 1","microtubule associated protein 1 light chain 3 alpha","phosphatidylinositol glycan anchor biosynthesis class U","acyl-CoA thioesterase 8","adipocyte plasma membrane associated protein","CUGBP Elav-like family member 4","zinc finger protein 516","ADNP homeobox 2","ribosome binding factor A","ubiquitin specific peptidase 14","VAMP associated protein A","methyltransferase like 4","lipin 2","structural maintenance of chromosomes flexible hinge domain containing 1","myomesin 1","myosin light chain 12A","centrosomal protein 76","ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 5","centrosomal protein 192","RNA guanine-7 methyltransferase","SMAD family member 7","lipase G, endothelial type","laminin subunit alpha 1","ring finger protein 125","ankyrin repeat domain 12","DNA polymerase iota","mindbomb E3 ubiquitin protein ligase 1","RB binding protein 8, endonuclease","RIO kinase 3","cleavage stimulation factor subunit 2","matrix remodeling associated 5","V-set and immunoglobulin domain containing 1","proteasome 26S subunit, non-ATPase 10","autophagy related 4A cysteine peptidase","steroid sulfatase","transducin beta like 1 X-linked","G protein-coupled receptor 143","progesterone receptor membrane component 1","DNA polymerase alpha 1, catalytic subunit","midline 1","NFKB activating protein","nuclear transport factor 2 like export factor 2","guanylate cyclase 2F, retinal","ATPase Na+/K+ transporting family member beta 4","ALG13 UDP-N-acetylglucosaminyltransferase subunit","phosphoribosyl pyrophosphate synthetase 2","toll like receptor 8","motile sperm domain containing 1","AMMECR nuclear protein 1","WD repeat domain 13","suppressor of variegation 3-9 homolog 1","X-linked inhibitor of apoptosis","stromal antigen 2","ATPase phospholipid transporting 11C","ATP binding cassette subfamily D member 1","coiled-coil domain containing 22","calcium voltage-gated channel subunit alpha1 F","synaptophysin","proteolipid protein 2","BMX non-receptor tyrosine kinase","plastin 3","N(alpha)-acetyltransferase 10, NatA catalytic subunit","renin binding protein","E74 like ETS transcription factor 4","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 1","myotubularin related protein 8","ankyrin repeat and SOCS box containing 9","zinc finger CCCH-type containing 12B","RB binding protein 7, chromatin remodeling factor","potassium voltage-gated channel subfamily D member 1","solute carrier family 25 member 14","FMRP translational regulator 1","Pim-2 proto-oncogene, serine/threonine kinase","solute carrier family 35 member A2","polyglutamine binding protein 1","retinoschisin 1","proprotein convertase subtilisin/kexin type 1 inhibitor","emerin","tafazzin","phosphoglycerate kinase 1","magnesium transporter 1","spermine synthase","phosphate regulating endopeptidase homolog X-linked","ubiquitin like 4A","CD99 molecule like 2","early endosome antigen 1","RP2 activator of ARL3 GTPase","jade family PHD finger 3","cyclin dependent kinase 16","ubiquitin specific peptidase 11","phosphate cytidylyltransferase 1, choline, beta","HIV-1 Tat specific factor 1","vestigial like family member 1","CD40 ligand","TIMP metallopeptidase inhibitor 1","gamma-aminobutyric acid type A receptor epsilon subunit","protocadherin 11 X-linked","FYVE, RhoGEF and PH domain containing 1","peptidylprolyl cis/trans isomerase, NIMA-interacting 4","porcupine O-acyltransferase","inter-alpha-trypsin inhibitor heavy chain family member 6","MAGE family member D2","RNA binding motif protein 3","Kruppel like factor 8","sushi repeat containing protein X-linked 2","synaptotagmin like 4","zinc finger DHHC-type containing 15","centromere protein I","polysaccharide biosynthesis domain containing 1","galactosidase alpha","armadillo repeat containing X-linked 3","brain expressed X-linked 4","rubicon like autophagy enhancer","sodium leak channel, non-selective","fibroblast growth factor 14","Nedd4 family interacting protein 2","TNF superfamily member 13b","fibronectin type III domain containing 3A","cytidine and dCMP deaminase domain containing 1","calcium binding protein 39 like","Kruppel like factor 5","serine/threonine kinase 24","acid phosphatase 5, tartrate resistant","DnaJ heat shock protein family (Hsp40) member C3","UDP-glucose glycoprotein glucosyltransferase 2","Rho guanine nucleotide exchange factor 7","fibroblast growth factor 9","poly(ADP-ribose) polymerase family member 4","SPT20 homolog, SAGA complex component","mitochondrial ribosomal protein S31","solute carrier family 25 member 15","karyopherin subunit alpha 3","fms related tyrosine kinase 1","regulator of cell cycle","von Willebrand factor A domain containing 8","diacylglycerol kinase eta","katanin catalytic subunit A1 like 1","integrator complex subunit 6","aconitate decarboxylase 1","dehydrogenase/reductase 12","TSC22 domain family member 1","CLN5 intracellular trafficking protein","mesothelin","mahogunin ring finger 1","zinc finger protein 629","TNFRSF1A associated via death domain","heat shock transcription factor 4","coronin 1A","mitogen-activated protein kinase 3","glycerophosphodiester phosphodiesterase domain containing 3","engulfment and cell motility 3","phosphorylase kinase regulatory subunit beta","LYR motif containing 1","nuclear transport factor 2","nucleoporin 93","centromere protein T","translin associated factor X interacting protein 1","nuclear factor of activated T cells 5","lon peptidase 2, peroxisomal","NEDD4 binding protein 1","ADP ribosylation factor like GTPase 2 binding protein","plasmolipin","C-C motif chemokine ligand 22","dihydroorotate dehydrogenase (quinone)","C-C motif chemokine ligand 17","CCCTC-binding factor","ACD shelterin complex subunit and telomerase recruitment factor","RNA polymerase II subunit C","par-6 family cell polarity regulator alpha","zinc finger protein 821","matrix metallopeptidase 15","U6 snRNA biogenesis phosphodiesterase 1","cytochrome b5 type B","coiled-coil domain containing 113","NME/NM23 nucleoside diphosphate kinase 3","NDRG family member 4","proteasome 26S subunit, non-ATPase 7","SET domain containing 6, protein lysine methyltransferase","solute carrier family 38 member 7","VAC14 component of PIKFYVE complex","hyaluronan synthase 3","transport and golgi organization 6 homolog","component of oligomeric golgi complex 4","sphingomyelin phosphodiesterase 3","solute carrier family 7 member 6 opposite strand","solute carrier family 7 member 6","phospholipase A2 group XV","epithelial splicing regulatory protein 2","fatty acid 2-hydroxylase","WD repeat domain 59","MON1 homolog B, secretory trafficking associated","C-X9-C motif containing 2","axin 1","host cell factor C1 regulator 1","NPR3 like, GATOR1 complex subunit","malonyl-CoA decarboxylase","N-methylpurine DNA glycosylase","N-terminal EF-hand calcium binding protein 2","hydroxysteroid dehydrogenase like 1","TATA-box binding protein associated factor, RNA polymerase I subunit C","N-acetylglucosamine-1-phosphodiester alpha-N-acetylglucosaminidase","WAP four-disulfide core domain 1","SEC14 like lipid binding 5","coactosin like F-actin binding protein 1","ubiquitin specific peptidase 10","cysteine rich secretory protein LCCL domain containing 2","TSC complex subunit 2","zinc finger protein 500","NME/NM23 nucleoside diphosphate kinase 4","ATP binding cassette subfamily C member 1","NODAL modulator 3","lipase maturation factor 1","cytosolic iron-sulfur assembly component 3","methenyltetrahydrofolate synthetase domain containing","chloride voltage-gated channel 7","hydroxyacylglutathione hydrolase like","adenine nucleotide translocase lysine methyltransferase","solute carrier family 7 member 5","meteorin, glial cell differentiation regulator","F-box protein 31","STIP1 homology and U-box containing protein 1","rhomboid like 1","nucleotide binding protein 1","ubiquitin conjugating enzyme E2 I","zona pellucida glycoprotein 2","MEFV innate immuity regulator, pyrin","crystallin mu","eukaryotic elongation factor 2 kinase","calpain 15","piezo type mechanosensitive ion channel component 1","G1 to S phase transition 1","zinc finger protein 174","clusterin associated protein 1","ubiquitin family domain containing 1","glutamyl-tRNA synthetase 2, mitochondrial","elongin B","golgi associated, gamma adaptin ear containing, ARF binding protein 2","calcineurin like phosphoesterase domain containing 1","ubiquitin specific peptidase 31","heme oxygenase 2","DnaJ heat shock protein family (Hsp40) member A3","CORO7-PAM16 readthrough","bifunctional apoptosis regulator","TOX high mobility group box family member 3","RB transcriptional corepressor like 2","quinolinate phosphoribosyltransferase","xylosyltransferase 1","PYD and CARD domain containing","RPGRIP1 like","MYC associated zinc finger protein","syntaxin 4","CDP-diacylglycerol--inositol 3-phosphatidyltransferase","branched chain keto acid dehydrogenase kinase","lysine acetyltransferase 8","NODAL modulator 1","interleukin 21 receptor","synaptotagmin 17","transmembrane channel like 5","centriolar coiled-coil protein 110","VPS35 endosomal protein sorting factor like","ring finger protein 40","lysine rich nucleolar protein 1","aquaporin 9","alpha and gamma adaptin binding protein","IQ motif containing H","lactamase beta","coronin 2B","C-terminal Src kinase","HECT and RLD domain containing E3 ubiquitin protein ligase family member 1","thyroid hormone receptor interactor 4","mitochondrial methionyl-tRNA formyltransferase","adaptor related protein complex 3 subunit beta 2","acyl-CoA synthetase bubblegum family member 1","RAB11A, member RAS oncogene family","cathepsin H","tetratricopeptide repeat domain 23","CD276 molecule","fumarylacetoacetate hydrolase","cell migration inducing hyaluronidase 1","RNA polymerase II associated protein 1","homer scaffold protein 2","EH domain containing 4","transmembrane protein 87A","zinc finger protein 106","centrosomal protein 152","ATPase phospholipid transporting 8B4 (putative)","DTW domain containing 1","GA binding protein transcription factor subunit beta 1","tight junction protein 1","Bcl2 modifying factor","Dmx like 2","secretogranin III","DnaJ heat shock protein family (Hsp40) member C17","eukaryotic translation initiation factor 3 subunit J","SPG11 vesicle trafficking associated, spatacsin","ras homolog family member V","VPS18 core subunit of CORVET and HOPS complexes","Opa interacting protein 5","solute carrier family 30 member 4","biogenesis of lysosomal organelles complex 1 subunit 6","myelin expression factor 2","serum/glucocorticoid regulated kinase family member 3","platelet derived growth factor receptor like","centrosome and spindle pole associated protein 1","zinc finger DHHC-type palmitoyltransferase 2","BRF2 RNA polymerase III transcription initiation factor subunit","tripartite motif containing 35","zinc finger AN1-type containing 1","RP1 axonemal microtubule associated","carbonic anhydrase 2","frizzled class receptor 3","integrator complex subunit 9","receptor interacting serine/threonine kinase 2","EYA transcriptional coactivator and phosphatase 1","nibrin","transient receptor potential cation channel subfamily A member 1","carboxypeptidase Q","2,4-dienoyl-CoA reductase 1","calbindin 1","inositol monophosphatase domain containing 1","secreted frizzled related protein 1","lysosomal protein transmembrane 4 beta","ubiquitin conjugating enzyme E2 W","POP1 homolog, ribonuclease P/MRP subunit","NIPA like domain containing 2","inhibitor of nuclear factor kappa B kinase subunit beta","plasminogen activator, tissue type","junctophilin 1","serine/threonine kinase 3","ganglioside induced differentiation associated protein 1","RAB2A, member RAS oncogene family","eukaryotic translation initiation factor 3 subunit E","ER membrane protein complex subunit 2","epithelial splicing regulatory protein 1","cellular communication network factor 4","N-myc downstream regulated 1","zinc finger C2HC-type containing 1A","interleukin 7","armadillo repeat containing 1","transcriptional repressor GATA binding 1","sperm associated antigen 1","chromatin accessibility complex subunit 1","neurocalcin delta","sorting nexin 16","ubiquitin protein ligase E3 component n-recognin 5","gasdermin D","tissue specific transplantation antigen P35B","pyrroline-5-carboxylate reductase 3","eukaryotic translation elongation factor 1 delta","annexin A13","squalene epoxidase","SH2 domain containing 4A","integrator complex subunit 10","exoribonuclease 1","solute carrier family 39 member 14","myotubularin related protein 9","leptin receptor overlapping transcript like 1","dynactin subunit 6","R3H domain and coiled-coil containing 1","glutathione-disulfide reductase","TNF receptor superfamily member 10a","UBX domain protein 8","protein phosphatase 2 catalytic subunit beta","glutamate rich 1","tumor suppressor candidate 3","Rho guanine nucleotide exchange factor 10","kelch domain containing 4","minichromosome maintenance complex component 4","potassium channel tetramerization domain containing 9","N-acylsphingosine amidohydrolase 1","BCL2 interacting protein 3 like","mannosidase alpha class 2B member 1","potassium calcium-activated channel subfamily N member 4","TUB like protein 2","nucleobindin 1","glycogen synthase 1","mitogen-activated protein kinase kinase kinase kinase 1","enoyl-CoA hydratase 1","heterogeneous nuclear ribonucleoprotein L","NFKB inhibitor beta","tubulin beta 4A class IVa","seryl-tRNA synthetase 2, mitochondrial","potassium voltage-gated channel subfamily A member 7","small nuclear ribonucleoprotein U1 subunit 70","CLPTM1 regulator of GABA type A receptor forward trafficking","RELB proto-oncogene, NF-kB subunit","CLK4 associating serine/arginine rich protein","lin-7 homolog B, crumbs cell polarity complex component","protein phosphatase 1 regulatory subunit 37","Fc fragment of IgG receptor and transporter","PIH1 domain containing 1","Rho/Rac guanine nucleotide exchange factor 18","protein phosphatase 1 regulatory subunit 13 like","peroxisomal biogenesis factor 11 gamma","ERCC excision repair 2, TFIIH core complex helicase subunit","DOT1 like histone lysine methyltransferase","pleckstrin homology domain containing J1","solute carrier family 17 member 7","ribonuclease H2 subunit A","kinesin light chain 3","CD37 molecule","splicing factor 3a subunit 2","anti-Mullerian hormone","LYL1 basic helix-loop-helix family member","ornithine decarboxylase antizyme 1","tRNA methyltransferase 1","syntaxin 10","Fc fragment of IgE receptor II","DM1 protein kinase","TBC1 domain family member 17","interleukin 4 induced 1","coiled-coil domain containing 130","PTOV1 extended AT-hook containing adaptor protein","TLE family member 5, transcriptional modulator","NOVA alternative splicing regulator 2","small glutamine rich tetratricopeptide repeat containing alpha","leukocyte immunoglobulin like receptor B1","mediator complex subunit 25","leukocyte immunoglobulin like receptor A1","small nuclear RNA activating complex polypeptide 2","chromosome 19 open reading frame 53","translocase of inner mitochondrial membrane 44","coiled-coil domain containing 61","interleukin 27 receptor subunit alpha","anti-silencing function 1B histone chaperone","troponin T1, slow skeletal type","VRK serine/threonine kinase 3","family with sequence similarity 32 member A","protein phosphatase 6 regulatory subunit 1","chromosome 19 open reading frame 44","mediator complex subunit 26","olfactomedin 2","RAS protein activator like 3","A-kinase anchoring protein 8","epoxide hydrolase 3","ilvB acetolactate synthase like","zinc finger protein 419","caspase 14","solute carrier family 1 member 6","aurora kinase C","POP4 homolog, ribonuclease P/MRP subunit","cyclin E1","URI1 prefoldin like chaperone","programmed cell death 5","ankyrin repeat domain 27","ribosomal protein S16","translocase of inner mitochondrial membrane 50","fibrillarin","dual specificity tyrosine phosphorylation regulated kinase 1B","glucose-6-phosphate isomerase","AKT serine/threonine kinase 2","phospholipase D family member 3","periaxin","protein inhibitor of activated STAT 4","NUMB like endocytic adaptor protein","Epstein-Barr virus induced 3","YJU2 splicing factor homolog","tubulin folding cofactor B","fibronectin type III and SPRY domain containing 1","RNA polymerase II subunit I","CAP-Gly domain containing linker protein 3","zinc finger RNA binding protein 2","solute carrier family 1 member 5","protein kinase D2","tight junction protein 3","amyloid beta precursor like protein 1","cactin, spliceosome C complex subunit","coiled-coil domain containing 9","heterogeneous nuclear ribonucleoprotein U like 1","fizzy and cell division cycle 20 related 1","BCL2 binding component 3","transforming growth factor beta 1","DENN domain containing 3","distal membrane arm assembly complex 2","CEA cell adhesion molecule 4","perilipin 3","myosin heavy chain 14","mitochondrial ribosomal protein L4","sialic acid binding Ig like lectin 8","CD79a molecule","intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)","ribosomal protein S19","NOP53 ribosome biogenesis factor","natural killer cell granule protein 7","intercellular adhesion molecule 5","electron transfer flavoprotein subunit beta","CD33 molecule","CEA cell adhesion molecule 5","cone-rod homeobox","BRISC and BRCA1 A complex member 1","tyrosine kinase 2","sulfotransferase family 2A member 1","cell division cycle 37","NSF attachment protein alpha","Rab acceptor 1","ATPase Na+/K+ transporting subunit alpha 3","Meis homeobox 3","protein tyrosine phosphatase receptor type S","cornifelin","multiple EGF like domains 8","KDEL endoplasmic reticulum protein retention receptor 1","cytohesin 2","glutamate rich WD repeat containing 1","glutamate ionotropic receptor NMDA type subunit 2D","coiled-coil domain containing 114","caspase recruitment domain family member 8","DNA ligase 1","sialic acid binding Ig like lectin 6","zinc finger protein 175","phospholipase A2 group IVC","sialic acid binding Ig like lectin 5","hyaluronan synthase 1","RAB3D, member RAS oncogene family","D-box binding PAR bZIP transcription factor","transmembrane protein 205","calcyphosine","phospholipid phosphatase related 2","family with sequence similarity 83 member E","Ras interacting protein 1","branched chain amino acid transaminase 2","MIER family member 2","pleckstrin homology domain containing A4","protein phosphatase 2 scaffold subunit Aalpha","transportin 2","WD repeat domain 83 opposite strand","glutaryl-CoA dehydrogenase","leukocyte immunoglobulin like receptor B5","deoxyribonuclease 2, lysosomal","microtubule associated serine/threonine kinase 1","leukocyte receptor cluster member 1","pre-mRNA processing factor 31","TCF3 fusion partner","Janus kinase 3","ribosomal protein L18a","solute carrier family 5 member 5","arrestin domain containing 2","phosphoinositide-3-kinase regulatory subunit 2","RAB3A, member RAS oncogene family","phosphodiesterase 4C","inositol-3-phosphate synthase 1","elongation factor for RNA polymerase II","CREB regulated transcription coactivator 1","cartilage oligomeric matrix protein","coatomer protein complex subunit epsilon","DEAD-box helicase 49","armadillo repeat containing 6","transmembrane protein 147","glyceraldehyde-3-phosphate dehydrogenase, spermatogenic","hepcidin antimicrobial peptide","upstream transcription factor 2, c-fos interacting","lipolysis stimulated lipoprotein receptor","KxDL motif containing 1","FKBP prolyl isomerase 8","SURP and G-patch domain containing 1","hepsin","zinc finger protein 14","sodium voltage-gated channel beta subunit 1","PBX homeobox 4","ETS2 repressor factor","glycogen synthase kinase 3 alpha","ATPase 13A1","zinc finger protein 574","signal induced proliferation associated 1 like 3","zinc finger protein 85","ETHE1 persulfide dioxygenase","cell adhesion molecule 4","SMG9 nonsense mediated mRNA decay factor","AVL9 cell migration associated","RUN domain containing 3B","cilia and flagella associated protein 69","GTP binding protein 10","RAS p21 protein activator 4","cyclin dependent kinase 6","peptidase, mitochondrial processing beta subunit","DnaJ heat shock protein family (Hsp40) member C2","tissue factor pathway inhibitor 2","Bet1 golgi vesicular membrane trafficking protein","nicotinamide phosphoribosyltransferase","TWIST neighbor","phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma","paraoxonase 3","paraoxonase 2","integrin subunit beta 8","HMG-box transcription factor 1","dihydrouridine synthase 4 like","Sp4 transcription factor","WD repeat domain 91","dynein axonemal heavy chain 11","Cbl proto-oncogene like 1","myotrophin","pleiotrophin","membrane palmitoylated protein 6","gasdermin E","ATPase H+ transporting V0 subunit a4","zinc finger CCCH-type containing, antiviral 1","tetratricopeptide repeat domain 26","oxoglutarate dehydrogenase","ArfGAP with dual PH domains 1","transcription factor EC","H2A.Z variant histone 2","caveolin 2","MET proto-oncogene, receptor tyrosine kinase","ring finger protein 32","limb development membrane protein 1","DnaJ heat shock protein family (Hsp40) member B6","LFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase","BRCA1 associated ATM activator 1","IQ motif containing E","vasoactive intestinal peptide receptor 2","tetraspanin 12","single stranded DNA binding protein 1","cadherin like and PC-esterase domain containing 1","3-hydroxyisobutyrate dehydrogenase","Tax1 binding protein 1","carboxypeptidase vitellogenic like","chimerin 2","growth factor receptor bound protein 10","abhydrolase domain containing 11","cordon-bleu WH2 repeat protein","FKBP prolyl isomerase 14","pleckstrin homology domain containing A8","syntaxin 1A","nucleotide binding oligomerization domain containing 1","glycyl-tRNA synthetase 1","corticotropin releasing hormone receptor 2","EPH receptor B6","MINDY lysine 48 deubiquitinase 4","caspase 2","coiled-coil-helix-coiled-coil-helix domain containing 2","heat shock protein family B (small) member 1","PDGFA associated protein 1","BUD31 homolog","pentatricopeptide repeat domain 1","cytochrome P450 family 3 subfamily A member 5","zinc finger with KRAB and SCAN domains 1","eukaryotic translation initiation factor 3 subunit B","sorting nexin 8","nudix hydrolase 1","protein tyrosine phosphatase receptor type Z1","TATA-box binding protein associated factor 6","WASP like actin nucleation promoting factor","hyaluronidase 4","aminoacyl tRNA synthetase complex interacting multifunctional protein 2","transferrin receptor 2","motile sperm domain containing 3","procollagen C-endopeptidase enhancer","F-box protein 24","RNA binding motif protein 28","ubiquitin specific peptidase 42","inosine monophosphate dehydrogenase 1","ArfGAP with FG repeats 2","LSM5 homolog, U6 small nuclear RNA and mRNA degradation associated","serpin family E member 1","adaptor related protein complex 1 subunit sigma 1","monoacylglycerol O-acyltransferase 3","core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase 1","procollagen-lysine,2-oxoglutarate 5-dioxygenase 3","replication protein A3","zinc finger HIT-type containing 1","claudin 15","glucocorticoid induced 1","PHD finger protein 14","nuclear respiratory factor 1","transmembrane protein 106B","enhancer of zeste 2 polycomb repressive complex 2 subunit","centrosomal protein 41","zinc finger protein 862","secreted frizzled related protein 4","mesoderm specific transcript","ankyrin repeat and MYND domain containing 2","actin related protein 3C","tetraspanin 13","retinoic acid receptor responder 2","anterior gradient 2, protein disulphide isomerase family member","aryl hydrocarbon receptor","coiled-coil-helix-coiled-coil-helix domain containing 3","GTPase, IMAP family member 2","transmembrane protein 176B","GLI family zinc finger 3","proteasome 20S subunit alpha 2","mitochondrial ribosomal protein L32","cytochrome c oxidase assembly factor 1 homolog","biliverdin reductase A","upregulator of cell proliferation","transmembrane protein 248","Ras homolog, mTORC1 binding","protein kinase AMP-activated non-catalytic subunit gamma 2","AE binding protein 1","DNA polymerase delta 2, accessory subunit","glucokinase","BAF chromatin remodeling complex subunit BCL7B","YKT6 v-SNARE homolog","transducin beta like 2","CAP-Gly domain containing linker protein 2","eukaryotic translation initiation factor 4H","LIM domain kinase 1","spermatogenesis associated 6 like","solute carrier family 1 member 1","fukutin","fibronectin type III and SPRY domain containing 1 like","contactin associated protein like 3","spindlin 1","nicotinamide riboside kinase 1","transmembrane protein 245","prune homolog 2 with BCH domain","multiple EGF like domains 9","tripartite motif containing 14","coronin 2A","transforming growth factor beta receptor 1","SEC61 translocon beta subunit","complement C5","osteoglycin","asporin","extracellular matrix protein 2","TLE family member 4, transcriptional corepressor","LIM homeobox 6","prostaglandin reductase 1","sushi domain containing 1","AT-hook transcription factor","TNF superfamily member 8","dynamin 1","endoglin","adenylate kinase 1","cell division cycle 37 like 1","plasminogen receptor with a C-terminal lysine","TBC1 domain family member 13","RIC1 homolog, RAB6A GEF complex partner 1","lysine demethylase 4C","dedicator of cytokinesis 8","KN motif and ankyrin repeat domains 1","ELAV like RNA binding protein 2","neuronal calcium sensor 1","testis associated actin remodelling kinase 1","potassium sodium-activated channel subfamily T member 1","far upstream element binding protein 3","cAMP responsive element binding protein 3","RGP1 homolog, RAB6A GEF complex partner 1","multiple PDZ domain crumbs cell polarity complex component","DExD/H-box helicase 58","endothelial differentiation related factor 1","phosphatidylinositol-4-phosphate 5-kinase type 1 beta","GLIS family zinc finger 3","BCL2 associated athanogene 1","Rap guanine nucleotide exchange factor 1","neural proliferation, differentiation and control 1","amyloid beta precursor protein binding family A member 1","senataxin","prostaglandin D2 synthase","ATP binding cassette subfamily A member 2","SH2 domain containing adaptor protein B","ubiquitin conjugating enzyme E2 R2","abhydrolase domain containing 17B","exosome component 3","zinc finger AN1-type containing 5","dishevelled segment polarity protein 1","PDZ and LIM domain 1","cyclin J","GATA binding protein 3","HPS1 biogenesis of lysosomal organelles complex 3 subunit 1","phytanoyl-CoA 2-hydroxylase","Ras association domain family member 4","dynamin binding protein","RAB11 family interacting protein 2","C-X-C motif chemokine ligand 12","ER lipid raft associated 1","eukaryotic translation initiation factor 3 subunit A","cubilin","tRNA aspartic acid methyltransferase 1","DExD-box helicase 50","mitogen-activated protein kinase 8","SEC23 interacting protein","arginyltransferase 1","NSE4 homolog A, SMC5-SMC6 complex component","pleckstrin homology domain containing A1","phosphatase domain containing paladin 1","unc-5 netrin receptor B","cadherin related 23","V-set immunoregulatory receptor","SPARC (osteonectin), cwcv and kazal like domains proteoglycan 2","mitochondrial calcium uptake 1","protein phosphatase 3 catalytic subunit beta","coiled-coil serine rich protein 2","bone morphogenetic protein receptor type 1A","multiple inositol-polyphosphate phosphatase 1","actin alpha 2, smooth muscle","lipase A, lysosomal acid type","twinkle mtDNA helicase","leucine zipper tumor suppressor 2","sideroflexin 3","Kazal type serine peptidase inhibitor domain 1","F-box and WD repeat domain containing 4","fibroblast growth factor 8","nucleophosmin/nucleoplasmin 3","tankyrase 2","golgi brefeldin A resistant guanine nucleotide exchange factor 1","Rho GTPase activating protein 21","cytoplasmic polyadenylation element binding protein 3","F-box and leucine rich repeat protein 15","CUE domain containing 2","SUFU negative regulator of hedgehog signaling","ankyrin repeat domain 26","acyl-CoA binding domain containing 5","phospholysine phosphohistidine inorganic pyrophosphate phosphatase","La ribonucleoprotein 4B","GTP binding protein 4","erythroid differentiation regulatory factor 1","BRCA2 and CDKN1A interacting protein","mitochondrial poly(A) polymerase","neuralized E3 ubiquitin protein ligase 1","SH3 and PX domains 2A","pitrilysin metallopeptidase 1","STN1 subunit of CST complex","mitogen-activated protein kinase kinase kinase 8","dickkopf WNT signaling pathway inhibitor 1","EBF transcription factor 3","glutaredoxin 3","sortilin related VPS10 domain containing receptor 1","transcription activation suppressor family member 2","X-prolyl aminopeptidase 1","structural maintenance of chromosomes 3","SHOC2 leucine rich repeat scaffold protein","transcription factor A, mitochondrial","coiled-coil domain containing 6","cullin 2","cyclin Y","ubiquitin conjugating enzyme E2 S","ribosomal protein L28","zinc finger MIZ-type containing 1","peptidylprolyl isomerase F","phenazine biosynthesis like protein domain containing","tetraspanin 14","leucine rich glioma inactivated 1","TBC1 domain family member 12","cytochrome P450 family 2 subfamily C member 18","keratin 23","crystallin beta A1","nuclear FMR1 interacting protein 2","GIT ArfGAP 1","ribosomal protein L19","F-box and leucine rich repeat protein 20","RUN domain containing 3A","upstream binding transcription factor","colony stimulating factor 3","proteasome 26S subunit, non-ATPase 3","CASC3 exon junction complex subunit","Rap guanine nucleotide exchange factor like 1","regulator of G protein signaling 9","ring finger protein 43","Wnt family member 3","aspartoacylase","RAD51 paralog C","septin 4","myotubularin related protein 4","tripartite motif containing 37","purinergic receptor P2X 1","DEAH-box helicase 40","tubulin delta 1","karyopherin subunit beta 1","golgi SNAP receptor complex member 2","pyridoxamine 5'-phosphate oxidase","ribosomal protein S6 kinase B1","tripartite motif containing 16 like","CDK5 regulatory subunit associated protein 3","chromobox 1","RecQ like helicase 5","phosphatidylinositol glycan anchor biosynthesis class L","galactokinase 1","integrator complex subunit 2","calmodulin binding transcription activator 2","mediator complex subunit 13","enolase 3","profilin 1","ring finger protein 167","solute carrier family 25 member 11","ras related dexamethasone induced 1","cholinergic receptor nicotinic epsilon subunit","retinoic acid induced 1","nucleoporin 88","complement C1q binding protein","solute carrier family 6 member 4","bleomycin hydrolase","carboxypeptidase D","golgi SNAP receptor complex member 1","coiled-coil domain containing 47","mediator complex subunit 31","developmentally regulated GTP binding protein 2","FtsJ RNA 2'-O-methyltransferase 3","A-kinase anchoring protein 10","aldehyde dehydrogenase 3 family member A1","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 2","intercellular adhesion molecule 2","synaptogyrin 2","B9 domain containing 1","UTP6 small subunit processome component","DEAD-box helicase 5","chromosome 17 open reading frame 75","cytohesin 1","proteasome 26S subunit, non-ATPase 11","galectin 3 binding protein","C-C motif chemokine ligand 2","C-C motif chemokine ligand 8","peroxisomal biogenesis factor 12","DExH-box helicase 58","lysine acetyltransferase 2A","RAB5C, member RAS oncogene family","N-acetyl-alpha-glucosaminidase","hydroxysteroid 17-beta dehydrogenase 1","MAX dimerization protein MLX","contactin associated protein 1","ABI family member 3","enhancer of zeste 1 polycomb repressive complex 2 subunit","distal-less homeobox 4","protein phosphatase 1 regulatory subunit 9B","collagen type I alpha 1 chain","mitochondrial ribosomal protein L27","vesicle amine transport 1","leucine rich repeat containing 59","Rho family GTPase 2","arachidonate 12-lipoxygenase, 12S type","histone deacetylase 5","ATP binding cassette subfamily C member 3","LUC7 like 3 pre-mRNA splicing factor","SMAD specific E3 ubiquitin protein ligase 2","dual specificity phosphatase 3","elongation factor Tu GTP binding domain containing 2","HLF transcription factor, PAR bZIP family member","solute carrier family 16 member 6","protein kinase cAMP-dependent type I regulatory subunit alpha","ephrin B3","FAM20A golgi associated secretory pathway pseudokinase","tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein epsilon","monocyte to macrophage differentiation associated","RAN guanine nucleotide release factor","diphthamide biosynthesis 1","mitogen-activated protein kinase kinase 6","dehydrogenase/reductase 7B","WD repeat and SOCS box containing 1","recoverin","SLC9A3 regulator 1","myosin heavy chain 3","N-acetyltransferase 9 (putative)","transmembrane protein 104","TNF alpha induced protein 1","intraflagellar transport 20","transmembrane protein 97","cerebellar degeneration related protein 2 like","peripheral myelin protein 22","unc-119 lipid binding chaperone","aldolase, fructose-bisphosphate C","SPT6 homolog, histone chaperone and transcription elongation factor","RAB34, member RAS oncogene family","PHD finger protein 12","transmembrane protein 33","gamma-aminobutyric acid type A receptor alpha4 subunit","SLAIN motif family member 2","OCIA domain containing 1","cell wall biogenesis 43 C-terminal homolog","defective in cullin neddylation 1 domain containing 4","ubiquitin specific peptidase 46","sulfotransferase family 1E member 1","cysteine rich hydrophobic domain 2","neuromedin U","capping protein inhibiting regulator of actin dynamics","late endosomal/lysosomal adaptor, MAPK and MTOR activator 3","nuclear factor kappa B subunit 1","amphiregulin","mannosidase beta","ubiquitin conjugating enzyme E2 D3","E74 like ETS transcription factor 2","NADH:ubiquinone oxidoreductase subunit C1","TBC1 domain family member 9","zinc finger protein 330","inositol polyphosphate-4-phosphatase type II B","GRB2 associated binding protein 1","kelch like family member 2","carboxypeptidase E","ribosomal protein L34","wolframin ER transmembrane glycoprotein","annexin A10","GrpE like 1, mitochondrial","GAR1 ribonucleoprotein","FSHD region gene 1","chloride voltage-gated channel 3","aminoadipate aminotransferase","polypeptide N-acetylgalactosaminyltransferase 7","DEAH-box helicase 15","Sep (O-phosphoserine) tRNA:Sec (selenocysteine) tRNA synthase","tripartite motif containing 2","solute carrier family 2 member 9","F-box and WD repeat domain containing 7","nei like DNA glycosylase 3","TBC1 domain family member 19","cytokine dependent hematopoietic cell linker","nuclear receptor binding SET domain protein 2","SH3 domain containing 19","stromal interaction molecule 2","major facilitator superfamily domain containing 10","glycine receptor beta","bone marrow stromal cell antigen 1","Rap guanine nucleotide exchange factor 2","HGF activator","sorting nexin 25","LRP2 binding protein","UFM1 specific peptidase 2","Kruppel like factor 3","kelch like family member 5","family with sequence similarity 149 member A","non-SMC condensin I complex subunit G","UDP-glucose 6-dehydrogenase","PPARG coactivator 1 alpha","DEAD-box helicase 25","HIV-1 Tat interactive protein 2","cathepsin C","coiled-coil domain containing 34","zinc finger and BTB domain containing 16","elongator acetyltransferase complex subunit 4","ZPR1 zinc finger","mitochondrial carrier 2","formin binding protein 4","tectorin alpha","sterol-C5-desaturase","cytotoxic and regulatory T cell molecule","junctional cadherin complex regulator","beta-1,3-glucuronyltransferase 1","heat shock protein family A (Hsp70) member 8","von Willebrand factor A domain containing 5A","DnaJ heat shock protein family (Hsp40) member C4","sialic acid acetylesterase","sorting nexin 15","leupaxin","deltex E3 ubiquitin ligase 4","autophagy related 2A","EH domain containing 1","oxysterol binding protein","unc-93 homolog B1, TLR signaling regulator","pseudouridine synthase 3","decapping enzyme, scavenger","lysine methyltransferase 5B","FAD dependent oxidoreductase domain containing 1","protein phosphatase 6 regulatory subunit 3","membrane spanning 4-domains A6A","membrane spanning 4-domains A4A","ST3 beta-galactoside alpha-2,3-sialyltransferase 4","carnitine palmitoyltransferase 1A","cyclin D1","coiled-coil domain containing 86","pre-mRNA processing factor 19","transmembrane protein 109","hemopexin","tripartite motif containing 3","cysteine and histidine rich domain containing 1","folate receptor alpha","anaphase promoting complex subunit 15","pannexin 1","Rho guanine nucleotide exchange factor 17","centrosomal protein 164","ring finger protein 141","centrosomal protein 126","eukaryotic translation initiation factor 4 gamma 2","interleukin 10 receptor subunit alpha","polypeptide N-acetylgalactosaminyltransferase 18","baculoviral IAP repeat containing 2","ubiquitination factor E4A","DEAD-box helicase 6","Cbl proto-oncogene","nectin cell adhesion molecule 1","homeodomain interacting protein kinase 3","KIAA1549 like","F-box protein 3","pyruvate dehydrogenase complex component X","solute carrier family 1 member 2","COMM domain containing 9","solute carrier family 15 member 3","CD5 molecule","1-aminocyclopropane-1-carboxylate synthase homolog (inactive)","midkine","autophagy and beclin 1 regulator 1","MAP kinase activating death domain","protein tyrosine phosphatase mitochondrial 1","N(alpha)-acetyltransferase 40, NatD catalytic subunit","cysteinyl-tRNA synthetase 1","solute carrier family 22 member 18","CD81 molecule","solute carrier family 35 member F2","chromosome 11 open reading frame 21","ELMO domain containing 1","SRY-box transcription factor 6","chromosome 11 open reading frame 58","phosphatidylinositol transfer protein membrane associated 1","ribosomal protein S13","aryl hydrocarbon receptor interacting protein","nucleoporin 98","NADH:ubiquinone oxidoreductase core subunit S8","T cell immune regulator 1, ATPase H+ transporting V0 subunit a3","choline kinase alpha","exophilin 5","HPS5 biogenesis of lysosomal organelles complex 2 subunit 2","general transcription factor IIH subunit 1","POU class 2 homeobox associating factor 1","protein tyrosine phosphatase non-receptor type 5","von Willebrand factor","proteasome 26S subunit, non-ATPase 9","prolyl 3-hydroxylase 3","PPFIA binding protein 1","pre-mRNA processing factor 40 homolog B","CD69 molecule","PR/SET domain 4","C-type lectin domain family 2 member B","coenzyme Q5, methyltransferase","selectin P ligand","coronin 1C","acid sensing ion channel subunit 1","caprin family member 2","tetraspanin 11","potassium channel tetramerization domain containing 10","solute carrier family 11 member 2","malectin","mevalonate kinase","cysteine and serine rich nuclear protein 2","calcium/calmodulin dependent protein kinase kinase 2","bridging integrator 2","interleukin 23 subunit alpha","ATP synthase F1 subunit beta","prostaglandin E synthase 3","BAF chromatin remodeling complex subunit BCL7A","arginine and serine rich coiled-coil 2","cytochrome P450 family 27 subfamily B member 1","lin-7 homolog A, crumbs cell polarity complex component","keratin 18","acyl-CoA synthetase short chain family member 3","tensin 2","GLI family zinc finger 1","protein phosphatase, Mg2+/Mn2+ dependent 1H","methionyl aminopeptidase 2","leukotriene A4 hydrolase","ETS transcription factor ELK3","solute carrier family 6 member 12","Wnt family member 5B","mago homolog B, exon junction complex subunit","transient receptor potential cation channel subfamily V member 4","integrin alpha FG-GAP repeat containing 2","forkhead box M1","proline rich protein HaeIII subfamily 1","protein arginine methyltransferase 8","poly(ADP-ribose) polymerase family member 11","actin related protein 2/3 complex subunit 3","GPN-loop GTPase 3","VPS29 retromer complex component","RAD51 associated protein 1","SH2B adaptor protein 3","A-kinase anchoring protein 3","MANSC domain containing 1","potassium voltage-gated channel subfamily A member 1","dual specificity phosphatase 16","cAMP responsive element binding protein like 2","acyl-CoA dehydrogenase family member 10","aldehyde dehydrogenase 2 family member","cyclin dependent kinase inhibitor 1B","G protein-coupled receptor class C group 5 member D","N(alpha)-acetyltransferase 25, NatB auxiliary subunit","germ cell associated 1","sodium channel epithelial 1 alpha subunit","lymphotoxin beta receptor","2-oxoglutarate and iron dependent oxygenase domain containing 2","cyclin dependent kinase 2 associated protein 1","2'-5'-oligoadenylate synthetase 3","2'-5'-oligoadenylate synthetase 2","ADP-ribosyltransferase 4 (Dombrock blood group)","matrix Gla protein","RAS protein activator like 1","Rho GDP dissociation inhibitor beta","general transcription factor IIH subunit 3","eukaryotic translation initiation factor 2B subunit alpha","DEAD-box helicase 55","solute carrier family 38 member 1","endonuclease, poly(U) specific","chromosome 12 open reading frame 49","replication factor C subunit 5","syntaxin 2","adhesion G protein-coupled receptor D1","coatomer protein complex subunit zeta 1","TBC1 domain family member 30","cullin associated and neddylation dissociated 1","interferon gamma","RAB5B, member RAS oncogene family","Mdm1 nuclear protein","nucleoporin 107","CCR4-NOT transcription complex subunit 2","timeless circadian regulator","cleavage and polyadenylation specific factor 6","KRR1 small subunit processome component homolog","mitochondrial ribosomal protein L51","glyceraldehyde-3-phosphate dehydrogenase","NOP2 nucleolar protein","chromodomain helicase DNA binding protein 4","acrosin binding protein","UHRF1 binding protein 1 like","COP9 signalosome subunit 7A","inhibitor of growth family member 4","G protein subunit beta 3","cell division cycle associated 3","choline phosphotransferase 1","ubiquitin specific peptidase 5","triosephosphate isomerase 1","N-acetylglucosamine-1-phosphate transferase subunits alpha and beta","splA/ryanodine receptor domain and SOCS box containing 2","enolase 2","atrophin 1","chromosome 12 open reading frame 57","protein tyrosine phosphatase non-receptor type 6","lysophosphatidylcholine acyltransferase 3","5'-nucleotidase domain containing 3","solute carrier organic anion transporter family member 1B3","Nanog homeobox","SDS3 homolog, SIN3A corepressor complex component","golgi transport 1B","protein kinase AMP-activated non-catalytic subunit beta 1","cytidine monophosphate N-acetylneuraminic acid synthetase","host cell factor C2","ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 1","C-type lectin domain family 4 member A","C2 calcium dependent domain containing 5","activation induced cytidine deaminase","RAB35, member RAS oncogene family","polyhomeotic homolog 1","cytochrome c oxidase subunit 6A1","regulatory factor X4","RIC8 guanine nucleotide exchange factor B","serine and arginine rich splicing factor 9","FGFR1 oncogene partner 2","killer cell lectin like receptor B1","collagen type XII alpha 1 chain","butyrophilin subfamily 3 member A3","tyrosyl-DNA phosphodiesterase 2","fyn related Src family tyrosine kinase","dermatan sulfate epimerase","RWD domain containing 1","radial spoke head component 4A","male germ cell associated kinase","transmembrane protein 14C","PAK1 interacting protein 1","glucosaminyl (N-acetyl) transferase 2 (I blood group)","small integral membrane protein 8","neural precursor cell expressed, developmentally down-regulated 9","centrosomal protein 85 like","androgen dependent TFPI regulating protein","anti-silencing function 1A histone chaperone","minichromosome maintenance 9 homologous recombination repair factor","family with sequence similarity 184 member A","RNA guanylyltransferase and 5'-phosphatase","mannosidase alpha class 1A member 1","gamma-aminobutyric acid type A receptor rho2 subunit","serine incorporator 1","HD domain containing 2","TPD52 like 1","histidine triad nucleotide binding protein 3","nuclear receptor coactivator 7","RHO family interacting cell polarization regulator 2","SAM and SH3 domain containing 1","uronyl 2-sulfotransferase","UL16 binding protein 1","F-box protein 5","mitochondrial translational release factor 1 like","peroxisome proliferator activated receptor delta","opioid receptor mu 1","FA complementation group E","TUB like protein 1","solute carrier family 26 member 8","mitogen-activated protein kinase 14","potassium channel tetramerization domain containing 20","serine/threonine kinase 38","serine and arginine rich splicing factor 3","superoxide dismutase 2","mitochondrial ribosomal protein L18","minichromosome maintenance complex component 3","ring finger protein 8","phosphatase and actin regulator 1","MAM domain containing glycosylphosphatidylinositol anchor 1","intestinal cell kinase","F-box protein 9","CD83 molecule","midasin AAA ATPase 1","SAYSVFN motif domain containing 1","BTB domain and CNC homolog 2","RNA binding motif protein 24","cyclase associated actin cytoskeleton regulatory protein 2","triggering receptor expressed on myeloid cells like 2","zinc finger protein 451","BCL2 associated athanogene 2","RAB23, member RAS oncogene family","G protein-coupled receptor 63","KH RNA binding domain containing, signal transduction associated 2","F-box and leucine rich repeat protein 4","cyclin C","E2F transcription factor 3","protein tyrosine phosphatase 4A1","activating signal cointegrator 1 complex subunit 3","blood vessel epicardial substance","mediator complex subunit 23","WASP family member 1","glycosylphosphatidylinositol specific phospholipase D1","aldehyde dehydrogenase 5 family member A1","crystallin beta-gamma domain containing 1","vanin 1","vanin 2","acyl-CoA thioesterase 13","small ArfGAP 1","ribosomal protein S12","chromosome 6 open reading frame 62","beta-1,3-glucuronyltransferase 2","geminin DNA replication inhibitor","EYA transcriptional coactivator and phosphatase 4","sine oculis binding protein homolog","nuclear receptor subfamily 2 group E member 1","sorting nexin 3","HBS1 like translational GTPase","tripartite motif containing 38","peroxisomal biogenesis factor 7","zinc finger and BTB domain containing 24","FIG4 phosphoinositide 5-phosphatase","p53 apoptosis effector related to PMP22","ARFGEF family member 3","solute carrier family 16 member 10","hdc homolog, cell cycle regulator","adhesion G protein-coupled receptor G6","phosphatase and actin regulator 2","EPM2A glucan phosphatase, laforin","solute carrier family 39 member 7","C-C motif chemokine receptor 6","solute carrier family 22 member 2","PHD finger protein 1","cutA divalent cation tolerance homolog","parkin coregulated","QKI, KH domain containing RNA binding","chromosome 6 open reading frame 118","phosphodiesterase 10A","MyoD family inhibitor","transcription factor EB","cyclin D3","bystin like","family with sequence similarity 120B","TATA-box binding protein","guanylate cyclase activator 1B","peripherin 2","BRD4 interacting chromatin remodeling complex associated protein like","protein phosphatase 2 regulatory subunit B'delta","mitochondrial ribosomal protein L2","protein tyrosine kinase 7 (inactive)","serum response factor","cullin 9","2'-deoxynucleoside 5'-phosphate N-hydrolase 1","dual specificity phosphatase 22","exocyst complex component 2","cytochrome c oxidase subunit 7A2","transmembrane protein 30A","GDP-mannose 4,6-dehydratase","SUMO specific peptidase 6","interphotoreceptor matrix proteoglycan 1","pre-mRNA processing factor 4B","TTK protein kinase","solute carrier family 29 member 1 (Augustine blood group)","cellular communication network factor 6","butyrophilin subfamily 2 member A1","laminin subunit alpha 4","terminal nucleotidyltransferase 5A","chloride intracellular channel 5","fibrosin like 1","ectonucleotide pyrophosphatase/phosphodiesterase family member 5","lymphocyte antigen 86","serine protease 16","T-box transcription factor 18","erbb2 interacting protein","protocadherin beta 2","histidyl-tRNA synthetase 2, mitochondrial","nudix hydrolase 12","centrosomal protein 72","mannosidase alpha class 2A member 1","semaphorin 5A","terminal nucleotidyltransferase 4A","growth hormone receptor","3-hydroxy-3-methylglutaryl-CoA synthase 1","death associated protein","NME/NM23 family member 5","bromodomain containing 8","kinesin family member 20A","nicotinamide nucleotide transhydrogenase","mitochondrial ribosomal protein S30","heat shock protein family A (Hsp70) member 9","mitochondrial ribosomal protein S27","prefoldin subunit 1","heparin binding EGF like growth factor","solute carrier family 4 member 9","lysyl oxidase","cadherin 9","amyloid beta precursor protein binding family B member 3","transmembrane and coiled-coil domains 6","secreted protein acidic and cysteine rich","IK cytokine","3-hydroxy-3-methylglutaryl-CoA reductase","ceramide transporter 1","Fas associated factor family member 2","phosphodiesterase 8B","CDC like kinase 4","protocadherin beta 15","hepatitis A virus cellular receptor 1","glutamate metabotropic receptor 6","IL2 inducible T cell kinase","ring finger protein 130","tRNA-histidine guanylyltransferase 1 like","arylsulfatase B","clathrin interactor 1","thrombospondin 4","CCR4-NOT transcription complex subunit 6","interleukin 12B","butyrophilin like 8","tetratricopeptide repeat domain 1","mutS homolog 3","Ras protein specific guanine nucleotide releasing factor 2","gamma-aminobutyric acid type A receptor gamma2 subunit","cyclin G1","RNA polymerase III subunit G","drosha ribonuclease III","cadherin 6","lamin B1","arrestin domain containing 3","golgi phosphoprotein 3","SUB1 regulator of transcription","natriuretic peptide receptor 3","family with sequence similarity 172 member A","threonyl-tRNA synthetase 1","leucyl and cystinyl aminopeptidase","phosphodiesterase 4D","RAD1 checkpoint DNA exonuclease","biogenesis of ribosomes BRX1","prolactin receptor","solute carrier family 12 member 7","RAD50 double strand break repair protein","ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 4","glucosamine-6-phosphate deaminase 1","protocadherin 12","S-phase kinase associated protein 1","nucleoporin 155","protein phosphatase 2 catalytic subunit alpha","fibroblast growth factor 1","nuclear receptor subfamily 3 group C member 1","chromosome 5 open reading frame 15","peptidylprolyl isomerase domain and WD repeat containing 1","LIF receptor subunit alpha","tripartite motif containing 23","trafficking protein particle complex 13","SEC24 homolog A, COPII coat complex component","thioredoxin domain containing 15","tetratricopeptide repeat domain 33","arginyl-tRNA synthetase 1","WW and C2 domain containing 1","macroH2A.1 histone","transcription elongation regulator 1","dihydropyrimidinase like 3","SMAD family member 5","casein kinase 1 alpha 1","HMG-box containing 3","endoplasmic reticulum-golgi intermediate compartment 1","platelet derived growth factor receptor beta","ATPase H+ transporting V0 subunit e1","BCL2 interacting protein 1","cytoplasmic polyadenylation element binding protein 4","histamine receptor H2","drebrin 1","zinc finger protein 346","unc-5 netrin receptor A","enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase","contactin 3","structural maintenance of chromosomes 4","selenoprotein K","actin related protein 8","TBCC domain containing 1","translocase of inner mitochondrial membrane domain containing 1","cereblon","histidine rich glycoprotein","BCL6 transcription repressor","homogentisate 1,2-dioxygenase","claudin 16","ADP ribosylation factor like GTPase 6","nephrocystin 3","CD86 molecule","angiomotin like 2","nitrilase family member 2","family with sequence similarity 162 member A","8-oxoguanine DNA glycosylase","karyopherin subunit alpha 1","propionyl-CoA carboxylase subunit beta","ubiquitin protein ligase E3A","armadillo repeat containing 8","centrosomal protein 70","retinol binding protein 2","retinol binding protein 1","solute carrier family 25 member 36","G protein-coupled receptor kinase 7","ring finger protein 7","transcription factor Dp-2","5'-3' exoribonuclease 1","lysine acetyltransferase 2B","serpin family I member 2","programmed cell death 10","Wnt family member 5A","6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4","collagen type VII alpha 1 chain","fibroblast growth factor 12","protein kinase cAMP-dependent type II regulatory subunit alpha","hes family bHLH transcription factor 1","ubiquitin specific peptidase 4","ArfGAP with coiled-coil, ankyrin repeat and PH domains 2","epithelial cell transforming 2","G protein subunit alpha transducin 1","G protein subunit alpha i2","trafficking from ER to golgi regulator","ubiquitin specific peptidase 9 Y-linked","hyaluronidase 1","tumor suppressor 2, mitochondrial calcium regulator","NPR2 like, GATOR1 complex subunit","ribosomal protein L24","cytochrome b561 family member D2","chromosome 3 open reading frame 14","FMR1 autosomal homolog 1","Cbl proto-oncogene B","BBX high mobility group box domain containing","intraflagellar transport 57","G protein subunit beta 4","HERV-H LTR-associating 2","IQ motif containing G","1,4-alpha-glucan branching enzyme 1","uridine monophosphate synthetase","nuclear cap binding protein subunit 2","sorting nexin 4","chromosome 3 open reading frame 52","FERM domain containing 4B","solute carrier family 41 member 3","rhophilin associated tail protein 1B","plexin A1","ATPase H+ transporting V1 subunit A","ankyrin repeat and BTB domain containing 1","podocalyxin like 2","uroplakin 1B","chondroitin sulfate proteoglycan 5","kelch like family member 18","SREBF chaperone","EF-hand and coiled-coil domain containing 1","KIAA1257","NIMA related kinase 11","mitochondrial ribosomal protein L3","phospholipid scramblase 4","HemK methyltransferase family member 1","cytokine inducible SH2 containing protein","MAPK activated protein kinase 3","activin A receptor type 2B","WD repeat domain 48","COMM domain containing 2","golgi reassembly stacking protein 1","peroxisomal biogenesis factor 5 like","ribosomal RNA processing 9, U3 small nucleolar RNA binding protein","ATP binding cassette subfamily C member 5","arylacetamide deacetylase","abhydrolase domain containing 14B","eukaryotic translation initiation factor 1B","ABHD14A-ACY1 readthrough","Rho guanine nucleotide exchange factor 26","kelch like family member 24","phospholipase C eta 1","vasoactive intestinal peptide receptor 1","dynein axonemal heavy chain 1","signal sequence receptor subunit 3","zinc finger and BTB domain containing 47","natural killer cell triggering receptor","chloride voltage-gated channel 2","forkhead box P1","eukaryotic translation initiation factor 4 gamma 1","signal peptidase complex subunit 1","NIMA related kinase 4","solute carrier family 4 member 3","INO80 complex subunit D","eukaryotic translation elongation factor 1 beta 2","deoxyguanosine kinase","MOB kinase activator 1A","KAT8 regulatory NSL complex subunit 3","lectin, mannose binding 2 like","rhotekin","tubulin tyrosine ligase","interleukin 1 alpha","C-C motif chemokine ligand 20","phosphoinositide kinase, FYVE-type zinc finger containing","potassium voltage-gated channel interacting protein 3","fumarylacetoacetate hydrolase domain containing 2A","nucleolin","actin related protein 1B","solute carrier family 35 member F5","zeta chain of T cell receptor associated protein kinase 70","actin related protein 3","STEAP3 metalloreductase","erythrocyte membrane protein band 4.1 like 5","transcription factor CP2 like 1","splicing factor 3b subunit 6","tumor protein p53 inducible protein 3","DnaJ heat shock protein family (Hsp40) member C27","signal transducing adaptor molecule 2","otoferlin","glycerol-3-phosphate dehydrogenase 2","cytohesin 1 interacting protein","activin A receptor type 1","tetratricopeptide repeat, ankyrin repeat and coiled-coil containing 1","solute carrier family 30 member 3","mitochondrial inner membrane protein MPV17","general transcription factor IIIC subunit 2","eukaryotic translation initiation factor 2B subunit delta","nuclear receptor binding protein 1","integrin subunit beta 6","integrin subunit alpha 4","proteasome 26S subunit, non-ATPase 14","sorting nexin 17","ankyrin repeat and SOCS box containing 3","protein phosphatase, Mg2+/Mn2+ dependent 1G","phosphodiesterase 1A","receptor accessory protein 6","proprotein convertase subtilisin/kexin type 4","APC regulator of WNT signaling pathway 2","interferon induced with helicase C domain 1","ribosomal protein S15","grancalcin","INO80 complex subunit B","mannosyl-oligosaccharide glucosidase","tetratricopeptide repeat domain 31","NADH:ubiquinone oxidoreductase core subunit S7","polycomb group ring finger 1","CAP-Gly domain containing linker protein family member 4","spectrin beta, non-erythrocytic 1","AUP1 lipid droplet regulating VLDL assembly factor","reticulon 4","HtrA serine peptidase 2","lysyl oxidase like 3","docking protein 1","polypeptide N-acetylgalactosaminyltransferase 3","DNA polymerase epsilon 4, accessory subunit","tachykinin receptor 1","coiled-coil domain containing 88A","acyl-CoA dehydrogenase long chain","eva-1 homolog A, regulator of programmed cell death","mitochondrial ribosomal protein L19","LanC like 1","WD repeat domain 75","EGF containing fibulin extracellular matrix protein 1","FA complementation group L","fibronectin 1","signal transducer and activator of transcription 1","glutaminase","poly(A) polymerase gamma","dynein axonemal heavy chain 6","peroxisomal trans-2-enoyl-CoA reductase","unc-50 inner nuclear membrane RNA binding protein","insulin like growth factor binding protein 2","ELMO domain containing 3","insulin like growth factor binding protein 5","ubiquitin specific peptidase 34","EF-hand domain family member D1","potassium inwardly rectifying channel subfamily J member 13","chaperonin containing TCP1 subunit 4","gamma-glutamyl carboxylase","EH domain binding protein 1","orthodenticle homeobox 1","thioredoxin domain containing 9","coenzyme Q10B","granulysin","splicing factor 3b subunit 1","ST3 beta-galactoside alpha-2,3-sialyltransferase 5","carbohydrate sulfotransferase 10","phosducin like 3","MOB family member 4, phocein","heat shock protein family E (Hsp10) member 1","lysine demethylase 3A","phospholipase C delta 4","charged multivesicular body protein 3","zinc finger protein 142","interleukin 1 receptor type 2","interleukin 1 receptor type 1","interleukin 1 receptor like 2","interleukin 1 receptor like 1","interleukin 18 receptor 1","interleukin 18 receptor accessory protein","solute carrier family 9 member A2","four and a half LIM domains 2","melanophilin","cyclin Pas1/PHO80 domain containing 1","UDP-glucuronate decarboxylase 1","ATP binding cassette subfamily B member 6 (Langereis blood group)","serine/threonine kinase 16","high density lipoprotein binding protein","protein phosphatase 1 regulatory subunit 7","PAS domain containing serine/threonine kinase","serine/threonine kinase 25","thyroid peroxidase","protein C, inactivator of coagulation factors Va and VIIIa","inhibitor of DNA binding 2","TATA-box binding protein associated factor, RNA polymerase I subunit B","hippocalcin like 1","ornithine decarboxylase 1","baculoviral IAP repeat containing 6","nucleolar protein 10","pleckstrin homology domain containing B2","golgi reassembly stacking protein 2","striatin","CCAAT enhancer binding protein zeta","protein kinase D3","DDB1 and CUL4 associated factor 17","glutaminyl-peptide cyclotransferase","RAB3 GTPase activating protein catalytic subunit 1","solute carrier family 25 member 12","regulator of microtubule dynamics 2","lactase","aspartyl-tRNA synthetase 1","serine and arginine rich splicing factor 7","syndecan 1","phospholipase C like 1 (inactive)","solute carrier family 1 member 4","SOS Ras/Rac guanine nucleotide exchange factor 1","kynureninase","WAS/WASL interacting protein family member 1","origin recognition complex subunit 2","cytochrome c oxidase subunit 7A2 like","partner of NOB1 homolog","origin recognition complex subunit 4","pleckstrin","Rho family GTPase 3","activating transcription factor 2","THADA armadillo repeat containing","AP2 associated kinase 1","trafficking kinesin protein 2","chromosome 2 open reading frame 42","TIA1 cytotoxic granule associated RNA binding protein","prenylcysteine oxidase 1","endothelial PAS domain protein 1","AT-rich interaction domain 3A","small ubiquitin like modifier 1","CD207 molecule","glutamate ionotropic receptor NMDA type subunit 3B","ATPase H+ transporting V1 subunit B1","nuclear factor, erythroid 2 like 2","mutS homolog 6","pleckstrin homology domain containing A3","sepiapterin reductase","EPH receptor A4","par-3 family cell polarity regulator beta","phenylalanyl-tRNA synthetase subunit beta","ALMS1 centrosome and basal body associated protein","BCL9 transcription coactivator","paired related homeobox 1","24-dehydrocholesterol reductase","DnaJ heat shock protein family (Hsp40) member C16","microtubule affinity regulating kinase 1","tenascin R","MORN repeat containing 1","glutathione peroxidase 7","calcyclin binding protein","sterol carrier protein 2","pappalysin 2","Ral GEF with PH domain and SH3 binding motif 2","angiopoietin like 1","centrosomal protein 104","FAM20B glycosaminoglycan xylosylkinase","transcription elongation factor A N-terminal and central domain containing 2","transmembrane protein 59","leucine rich repeat containing 42","WD repeat containing, antisense to TP73","mitochondrial ribosomal protein L37","isoprenylcysteine carboxyl methyltransferase","ribosomal protein L22","chromodomain helicase DNA binding protein 5","quiescin sulfhydryl oxidase 1","syntaxin binding protein 3","PHD finger protein 13","ERBB receptor feedback inhibitor 1","Parkinsonism associated deglycase","KIAA1324","opioid receptor delta 1","adenosine monophosphate deaminase 2","serine and arginine rich splicing factor 4","mitochondrial trans-2-enoyl-CoA reductase","potassium voltage-gated channel subfamily C member 4","ER degradation enhancing alpha-mannosidase like protein 3","WD repeat domain 77","ATP synthase peripheral stalk-membrane subunit b","RAP1A, member of RAS oncogene family","histone deacetylase 1","capping actin protein of muscle Z-line subunit alpha 1","S100P binding protein","ring finger protein 19B","secretory carrier membrane protein 3","tripartite motif containing 62","ASH1 like histone lysine methyltransferase","DLG associated protein 3","splicing factor proline and glutamine rich","ras homolog family member U","gon-4 like","Rho/Rac guanine nucleotide exchange factor 2","late endosomal/lysosomal adaptor, MAPK and MTOR activator 2","myocyte enhancer factor 2D","dedicator of cytokinesis 7","spermidine synthase","F-box protein 2","F-box protein 6","chromosome 1 open reading frame 21","SWT1 RNA endoribonuclease homolog","mitotic arrest deficient 2 like 2","DnaJ heat shock protein family (Hsp40) member C6","leptin receptor","influenza virus NS1A binding protein","KIAA2013","mitofusin 2","proteoglycan 4","migration and invasion inhibitory protein","SMG7 nonsense mediated mRNA decay factor","neutrophil cytosolic factor 2","phosducin","solute carrier family 35 member D1","phospholipase A2 group IVA","growth arrest and DNA damage inducible alpha","Wnt ligand secretion mediator","PR/SET domain 2","regulator of G protein signaling 2","Ro60, Y RNA binding protein","adenosine monophosphate deaminase 1","ubiquitin C-terminal hydrolase L5","BCAS2 pre-mRNA processing factor","serine and arginine rich splicing factor 11","cystathionine gamma-lyase","agmatinase","olfactomedin like 3","TNNI3 interacting kinase","complement factor H related 3","pleckstrin homology and RUN domain containing M2","crystallin zeta","putative homeodomain transcription factor 1","zinc finger and BTB domain containing 17","CD58 molecule","transcription factor AP-2 epsilon","CD2 molecule","transcription termination factor 2","nuclear receptor subfamily 5 group A member 2","kinesin family member 21B","transmembrane protein 9","ADP-ribosylhydrolase like 2","MAP7 domain containing 1","tryptophanyl tRNA synthetase 2, mitochondrial","organic solute carrier partner 1","mitochondrial ribosomal protein S15","exocyst complex component 8","glyceronephosphate O-acyltransferase","translin associated factor X","chromosome 1 open reading frame 109","Ras related GTP binding C","galectin 8","5-methyltetrahydrofolate-homocysteine methyltransferase","bone morphogenetic protein 8b","MYCL proto-oncogene, bHLH transcription factor","signal induced proliferation associated 1 like 2","rearranged L-myc fusion","kynurenine 3-monooxygenase","zinc finger protein 684","regulating synaptic membrane exocytosis 3","AKT serine/threonine kinase 3","ETS variant transcription factor 3","acyl-CoA dehydrogenase medium chain","signaling lymphocytic activation molecule family member 1","CD48 molecule","adhesion G protein-coupled receptor L2","peptidyl arginine deiminase 2","succinate dehydrogenase complex iron sulfur subunit B","microfibril associated protein 2","ribosome production factor 1 homolog","lysine demethylase 5B","UDP-N-acetylglucosamine pyrophosphorylase 1","chitobiase","regulator of G protein signaling 4","kelch like family member 12","SSX family member 2 interacting protein","zinc finger HIT-type containing 6","phospholipase A2 group IID","RB binding protein 5, histone lysine methyltransferase complex subunit","guanylate binding protein 3","guanylate binding protein 1","kinesin family member 17","G protein-coupled receptor 89A","cyclin dependent kinase 18","RAB29, member RAS oncogene family","CD160 molecule","endothelin converting enzyme 1","3-hydroxy-3-methylglutaryl-CoA lyase","UDP-galactose-4-epimerase","inhibitor of DNA binding 3, HLH protein","complement C3d receptor 2","CD46 molecule","pre-mRNA processing factor 3","aph-1 homolog A, gamma-secretase subunit","prolyl 3-hydroxylase 1","solute carrier family 2 member 1","EBNA1 binding protein 2","cell division cycle 20","MPL proto-oncogene, thrombopoietin receptor","importin 13","ATPase H+ transporting V0 subunit b","beta-1,4-galactosyltransferase 2","ERI1 exoribonuclease family member 3","patched 2","aldo-keto reductase family 1 member A1","peroxiredoxin 1","phosphoinositide-3-kinase regulatory subunit 3","tetraspanin 1","basic leucine zipper nuclear factor 1","coiled-coil domain containing 181","solute carrier family 19 member 2","fatty acid amide hydrolase","NOP2/Sun RNA methyltransferase 4","transmembrane p24 trafficking protein 5","maestro heat like repeat family member 9","down-regulator of transcription 1","calponin 3","proline rich coiled-coil 2C","coagulation factor III, tissue factor","ATP binding cassette subfamily D member 3","vesicle associated membrane protein 4","diphthamide biosynthesis 5","Fas ligand","polypyrimidine tract binding protein 2","TNF superfamily member 4","peroxiredoxin 6","aspartyl-tRNA synthetase 2, mitochondrial","hydroxysteroid 11-beta dehydrogenase 1","interferon regulatory factor 6","UTP25 small subunit processor component","serpin family C member 1","RCAN family member 3","SYF2 pre-mRNA splicing factor","arginine and serine rich protein 1","solute carrier family 35 member A3","REST corepressor 3","stathmin 1","mitochondrial fission regulator 1 like","mannosidase alpha class 1C member 1","NIMA related kinase 2","ribosomal protein S6 kinase A1","dehydrodolichyl diphosphate synthase subunit","neudesin neurotrophic factor","NSL1 component of MIS12 kinetochore complex","prospero homeobox 1","AT-rich interaction domain 1A","centromere protein F","replication protein A2","protein phosphatase 1 regulatory subunit 8","syntaxin 12","mitochondrial amidoxime reducing component 2","solute carrier family 5 member 9","oxysterol binding protein like 9","thioredoxin domain containing 12","extended synaptotagmin 2","CD3e molecule associated protein","mesoderm development LRP chaperone","reticulocalbin 2","cholinergic receptor nicotinic beta 4 subunit","mucin 5B, oligomeric mucus/gel-forming","cathepsin D","collectin subfamily member 11","stromal antigen 1","serine/threonine kinase 11","lysine methyltransferase 2A","intraflagellar transport 46","solute carrier family 8 member A2","kaptin, actin binding protein","ribosomal protein S25","kinesin family member 14","troponin T2, cardiac type","DEAD-box helicase 59","calmodulin regulated spectrin associated protein family member 2","activating transcription factor 6","melanoregulin","FAST kinase domains 2","neuropilin 2","cAMP responsive element binding protein 1","Kruppel like factor 7","transthyretin","beta-1,4-galactosyltransferase 6","chromosome 1 open reading frame 54","carbonic anhydrase 14","cancer susceptibility 1","lymphoid restricted membrane protein","ATPase phospholipid transporting 10B (putative)","signal peptidase complex subunit 2","ubiquitin specific peptidase 35","filamin A interacting protein 1","caspase 8 associated protein 2","high mobility group nucleosomal binding domain 3","ubiquitin protein ligase E3D","ankyrin repeat domain 13C","SH3GL interacting endocytic adaptor 1","PHD finger protein 3","androglobin","PLAG1 like zinc finger 1","F-box protein 30","TNF alpha induced protein 3","A-kinase anchoring protein 7","RAB32, member RAS oncogene family","MYB proto-oncogene, transcription factor","aldehyde dehydrogenase 8 family member A1","serum/glucocorticoid regulated kinase 1","ring finger protein 146","arginase 1","cellular communication network factor 2","polyamine modulated factor 1 binding protein 1","F-box and leucine rich repeat protein 5","mediator complex subunit 28","solute carrier family 16 member 7","ribitol xylosyltransferase 1","zinc finger protein 430","vesicle associated membrane protein 8","DNA cross-link repair 1B","myosin light chain 12B","forkhead box O3","armadillo repeat containing 2","ribophorin II","TGFB induced factor homeobox 2","calsequestrin 2","polycystin 2, transient receptor potential cation channel","ATP binding cassette subfamily G member 2 (Junior blood group)","secreted phosphoprotein 1","starch binding domain 1","cyclin I","retinoic acid receptor responder 1","major facilitator superfamily domain containing 1","RAB3 GTPase activating non-catalytic protein subunit 2","eukaryotic elongation factor 2 lysine methyltransferase","periplakin","ubinuclein 1","Kruppel like factor 12","ubiquitin C-terminal hydrolase L3","protocadherin 17","HCLS1 binding protein 3","lipid droplet associated hydrolase","WD repeat domain 35","cyclin D2","fibroblast growth factor 23","elongation factor for RNA polymerase II 2","dynein axonemal heavy chain 7","cytochrome P450 family 20 subfamily A member 1","NADH:ubiquinone oxidoreductase subunit B3","general transcription factor IIIC subunit 3","SATB homeobox 2","ubiquitin conjugating enzyme E2 B","transient receptor potential cation channel subfamily M member 6","guanine deaminase","Kruppel like factor 9","tight junction protein 2","ECRG4 augurin precursor","integrin subunit beta 1 binding protein 1","cleavage and polyadenylation specific factor 3","phosphatidylinositol glycan anchor biosynthesis class Z","SUMO specific peptidase 5","coiled-coil domain containing 92","chromosome 1 open reading frame 198","HEAT repeat containing 1","polypyrimidine tract binding protein 3","RAD23 homolog B, nucleotide excision repair protein","FKBP prolyl isomerase 15","catenin alpha like 1","actin binding transcription modulator","WD repeat domain 34","SET nuclear proto-oncogene","protein phosphatase 2 phosphatase activator","GLE1 RNA export mediator","RAB14, member RAS oncogene family","centriolin","tripartite motif containing 32","F-box and WD repeat domain containing 2","PHD finger protein 19","NIMA related kinase 6","B-box and SPRY domain containing","protein phosphatase 6 catalytic subunit","NADH:ubiquinone oxidoreductase subunit A8","haloacid dehalogenase like hydrolase domain containing 3","RNA binding motif protein 18","hydroxysteroid dehydrogenase like 2","MAPK associated protein 1","nuclear receptor subfamily 4 group A member 3","inversin","polypeptide N-acetylgalactosaminyltransferase 12","DENN domain containing 1A","ALG2 alpha-1,3/1,6-mannosyltransferase","colony stimulating factor 3 receptor","3-ketodihydrosphingosine reductase","vacuolar protein sorting 4 homolog B","chromosome 19 open reading frame 25","zinc finger and BTB domain containing 45","YLP motif containing 1","DDB1 and CUL4 associated factor 4","prospero homeobox 2","FCF1 rRNA-processing protein","placental growth factor","interferon alpha inducible protein 27 like 2","basal body orientation factor 1","NIMA related kinase 9","acylphosphatase 1","intraflagellar transport 43","NPC intracellular cholesterol transporter 2","dynein axonemal light chain 1","interferon regulatory factor 2 binding protein like","acyl-CoA thioesterase 2","latent transforming growth factor beta binding protein 2","apoptosis resistant E3 ubiquitin protein ligase 1","mutL homolog 3","tubulin tyrosine ligase like 5","feline leukemia virus subgroup C cellular receptor family member 2","ATP binding cassette subfamily D member 4","dihydrolipoamide S-succinyltransferase","protein phosphatase 4 regulatory subunit 4","transforming growth factor beta 3","zinc finger C2HC-type containing 1C","SRA stem-loop interacting RNA binding protein","RNA binding motif protein 25","aldehyde dehydrogenase 6 family member A1","G protein-coupled receptor 68","eukaryotic translation initiation factor 2B subunit beta","NRDE-2, necessary for RNA interference, domain containing","coenzyme Q6, monooxygenase","zinc finger protein 410","ras homolog family member Q","G protein-coupled receptor 75","SPT7 like, STAGA complex gamma subunit","kelch like family member 29","DNA methyltransferase 3 alpha","transmembrane protein 214","ATPase family AAA domain containing 2B","FKBP prolyl isomerase 1B","atlastin GTPase 2","yippee like 5","family with sequence similarity 98 member A","Yip1 domain family member 4","aftiphilin","galectin like","cannabinoid receptor interacting protein 1","BAF chromatin remodeling complex subunit BCL11A","CXXC repeat containing interactor of PDZ3 domain","epithelial cell adhesion molecule","solute carrier family 17 member 5","opioid growth factor receptor like 1","SMC5-SMC6 complex localization factor 2","insulin degrading enzyme","interferon induced protein with tetratricopeptide repeats 3","interferon induced protein with tetratricopeptide repeats 2","glycerol-3-phosphate acyltransferase, mitochondrial","cutC copper transporter","protein phosphatase 1 regulatory subunit 3C","pyridine nucleotide-disulphide oxidoreductase domain 2","cyclin and CBS domain divalent metal cation transport mediator 1","MAX interactor 1, dimerization protein","survival motor neuron domain containing 1","chromosome 10 open reading frame 88","helicase, lymphoid specific","prolactin releasing hormone receptor","tectonic family member 3","DENN domain containing 10","arginine vasopressin induced 1","WD repeat domain 11","armadillo like helical domain containing 3","potassium voltage-gated channel interacting protein 2","cilia and flagella associated protein 58","glutamic-oxaloacetic transaminase 1","chromosome 10 open reading frame 95","G protein subunit alpha 13","KAT8 regulatory NSL complex subunit 1","homeobox B3","dual specificity phosphatase 1","pantothenate kinase 3","RNA terminal phosphate cyclase like 1","caspase activity and apoptosis inhibitor 1","MOB kinase activator 3B","insulin like 6","insulin like 4","melan-A","CD274 molecule","nucleoporin 43","methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1 like","LDL receptor related protein 11","coiled-coil domain containing 170","protein-L-isoaspartate (D-aspartate) O-methyltransferase","pleckstrin homology and RhoGEF domain containing G1","MYC target 1","chromosome X open reading frame 21","cysteine rich transmembrane module containing 1","WD repeat domain 55","ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 3","protocadherin beta 14","mitochondrial ribosomal protein S14","centromere protein L","SEC16 homolog B, endoplasmic reticulum export factor","golgin, RAB6 interacting","acetyl-CoA acetyltransferase 2","t-complex 1","sorting nexin 19","potassium inwardly rectifying channel subfamily J member 5","Myb/SANT DNA binding domain containing 2","tumor protein p53 regulated apoptosis inducing protein 1","arrestin 3","PDZ domain containing 11","solute carrier family 10 member 7","NudC domain containing 1","ENY2 transcription and export complex 2 subunit","microtubule associated serine/threonine kinase like","KIAA1217","plexin domain containing 2","enhancer of polycomb homolog 1","IQ motif and Sec7 domain ArfGEF 3","coiled-coil domain containing 77","TATA-box binding protein associated factor 12","ecto-NOX disulfide-thiol exchanger 1","mitochondrial translation release factor 1","spermatogenesis and oogenesis specific basic helix-loop-helix 2","DnaJ heat shock protein family (Hsp40) member C15","proline and serine rich 1","ubiquitin fold modifier 1","WW domain binding protein 4","E74 like ETS transcription factor 1","SMAD family member 9","heat shock protein family H (Hsp110) member 1","kelch repeat and BTB domain containing 7","ALG5 dolichyl-phosphate beta-glucosyltransferase","exosome component 8","eukaryotic translation termination factor 1","transforming growth factor beta induced","family with sequence similarity 53 member C","SIL1 nucleotide exchange factor","poly(A) binding protein interacting protein 2","myotilin","lysine demethylase 3B","early growth response 1","stress associated endoplasmic reticulum protein 1","plastin 1","ZFP30 zinc finger protein","nuclear receptor subfamily 2 group C member 1","UTP20 small subunit processome component","thymopoietin","ADP ribosylation factor like GTPase 1","glycosyltransferase 8 domain containing 2","mitochondrial transcription termination factor 2","suppressor of cytokine signaling 2","nuclear transcription factor Y subunit beta","WASH complex subunit 3","apoptotic peptidase activating factor 1","dual specificity phosphatase 4","clusterin","TNF receptor superfamily member 10b","sorbin and SH3 domain containing 3","protein tyrosine kinase 2 beta","cholinergic receptor nicotinic alpha 2 subunit","protein phosphatase 3 catalytic subunit gamma","PDZ and LIM domain 2","epoxide hydrolase 2","ring finger protein 170","UbiA prenyltransferase domain containing 1","TAR DNA binding protein","TNF receptor superfamily member 8","zinc finger protein 706","lysophospholipase 1","cysteine rich secretory protein LCCL domain containing 1","COP9 signalosome subunit 5","retinol dehydrogenase 10","eosinophil peroxidase","A-kinase anchoring protein 1","coilin","tripartite motif containing 25","serine carboxypeptidase 1","speckle type BTB/POZ protein","solute carrier family 35 member B1","T-box transcription factor 4","testis expressed 14, intercellular bridge forming factor","family with sequence similarity 117 member A","non-SMC condensin I complex subunit H","lecithin retinol acyltransferase","transmembrane 131 like","meiotic nuclear divisions 1","tripartite motif containing 6","ATP binding cassette subfamily C member 11","terminal nucleotidyltransferase 4B","adenylate cyclase 7","centrosomal protein 89","teashirt zinc finger homeobox 3","enoyl-CoA hydratase domain containing 2","phospholipase B domain containing 1","proline rich protein BstNI subfamily 2","pyridine nucleotide-disulphide oxidoreductase domain 1","BCL2 like 14","paraspeckle component 1","zinc finger protein 549","alpha-1-B glycoprotein","zinc finger and SCAN domain containing 18","zinc finger protein 211","regulator of G protein signaling like 1","LIM homeobox 4","ring finger protein 2","tRNA methyltransferase 1 like","SEC22 homolog A, vesicle trafficking protein","cystatin A","developmental pluripotency associated 4","popeye domain containing 2","beta-1,4-galactosyltransferase 4","N(alpha)-acetyltransferase 50, NatE catalytic subunit","CD80 molecule","kinesin family member 18A","desumoylating isopeptidase 2","mitogen-activated protein kinase 8 interacting protein 1","cryptochrome circadian regulator 2","peroxisomal biogenesis factor 16","catalase","paired immunoglobin like type 2 receptor beta","zinc finger MYM-type containing 2","TBC1 domain family member 15","adhesion G protein-coupled receptor B2","zinc finger CCHC-type containing 17","fatty acid binding protein 3","KH RNA binding domain containing, signal transduction associated 1","transmembrane protein 39B","C-C motif chemokine receptor like 2","C-C motif chemokine receptor 2","RNA polymerase III subunit G like","growth hormone secretagogue receptor","TNF superfamily member 10","zinc finger protein 639","phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha","PDS5 cohesin associated factor A","transmembrane protein 156","lipoic acid synthetase","carboxypeptidase X, M14 family member 2","transmembrane protein 54","zinc finger and SCAN domain containing 20","CUB and Sushi multiple domains 2","hippocalcin","ligand dependent nuclear receptor interacting factor 1","chloride channel CLIC like 1","G protein signaling modulator 2","glycosyltransferase like domain containing 1","C-X-C motif chemokine receptor 4","zinc finger RANBP2-type containing 3","activin A receptor type 2A","DNA polymerase kappa","synaptic vesicle glycoprotein 2C","fms related tyrosine kinase 3","ribosomal protein L21","mitochondrial translational initiation factor 3","general transcription factor IIIA","RAS like family 11 member A","ubiquitin like 3","forty-two-three domain containing 1","mitochondrial transcription termination factor 4","SAM and SH3 domain containing 3","OCRL inositol polyphosphate-5-phosphatase","mitochondrial ribosomal protein S2","fibromodulin","lymphocyte transmembrane adaptor 1","plasminogen","KIAA1191","coatomer protein complex subunit alpha","CD244 molecule","lymphocyte antigen 9","heparan sulfate-glucosamine 3-sulfotransferase 2","RB binding protein 6, ubiquitin ligase","zinc finger CCCH-type containing 7A","serine active site containing 1","annexin A11","LIM domain binding 3","shieldin complex subunit 2","peroxiredoxin like 2A","zinc finger protein 205","N(alpha)-acetyltransferase 60, NatF catalytic subunit","ribosomal protein L5","outer dense fiber of sperm tails 2 like","prostaglandin F receptor","spermatogenesis associated 1","tRNA methyltransferase 13 homolog","leucine rich repeat containing 39","RWD domain containing 3","zinc finger protein 644","coiled-coil domain containing 18","RNA polymerase II associated protein 2","solute carrier family 66 member 2","Bardet-Biedl syndrome 9","PMS1 homolog 2, mismatch repair system component","zinc finger MIZ-type containing 2","septin 7","endonuclease/exonuclease/phosphatase family domain containing 1","kelch like family member 7","HERPUD family member 2","chromobox 3","heterogeneous nuclear ribonucleoprotein A2/B1","WAS/WASL interacting protein family member 3","family with sequence similarity 126 member A","inhibin subunit beta A","FKBP prolyl isomerase 9","5'-nucleotidase, cytosolic IIIA","ADP ribosylation factor like GTPase 4A","CCZ1 homolog, vacuolar protein trafficking and biogenesis associated","DNA polymerase mu","mitochondrial rRNA methyltransferase 2","SMU1 DNA replication regulator and spliceosomal factor","GLI pathogenesis related 2","solute carrier family 25 member 51","clathrin light chain A","reversion inducing cysteine rich protein with kazal motifs","aconitase 1","PHD finger protein 24","dynein axonemal intermediate chain 1","DDB1 and CUL4 associated factor 10","ciliary neurotrophic factor receptor","KIAA1549","tripartite motif containing 24","cell cycle regulator of NHEJ","caldesmon 1","aldo-keto reductase family 1 member D1","plasminogen activator, urokinase","serglycin","carbohydrate sulfotransferase 3","BicC family RNA binding protein 1","CDGSH iron sulfur domain 1","early growth response 2","ecdysoneless cell cycle regulator","prolyl 4-hydroxylase subunit alpha 1","solute carrier family 25 member 16","ZW10 interacting kinetochore protein","VPS26, retromer complex component A","RNA binding motif protein 19","citron rho-interacting serine/threonine kinase","intraflagellar transport 81","acyl-CoA dehydrogenase short chain","hydrogen voltage gated channel 1","DEAD-box helicase 54","mediator complex subunit 13L","cyclin dependent kinase inhibitor 2C","ring finger protein 11","Ras association domain family member 8","basic helix-loop-helix family member e41","sarcospan","inositol 1,4,5-trisphosphate receptor type 2","coiled-coil domain containing 91","N-terminal EF-hand calcium binding protein 1","WW domain containing E3 ubiquitin protein ligase 1","acyl-CoA thioesterase 9","peroxiredoxin 4","DExD-box helicase 39A","protein kinase N1","telomerase RNA component interacting RNase","adhesion G protein-coupled receptor E5","WD repeat domain 83","GIPC PDZ domain containing family member 1","SPRY domain containing 7","EBP like","ATPase copper transporting beta","zinc finger CCCH-type containing 13","neurolysin","centromere protein K","optineurin","inter-alpha-trypsin inhibitor heavy chain 5","activating transcription factor 1","Ts translation elongation factor, mitochondrial","Rho GTPase activating protein 9","NCK associated protein 1 like","matrix metallopeptidase 19","prefoldin subunit 5","spermatogenesis associated serine rich 2","ORMDL sphingolipid biosynthesis regulator 2","nuclear receptor subfamily 4 group A member 1","phosphodiesterase 1B","cyclin dependent kinase 2","LDL receptor related protein 1","autophagy related 101","nuclear factor, erythroid 2","IKAROS family zinc finger 4","single-strand-selective monofunctional uracil-DNA glycosylase 1","tubulin alpha 1b","EEF1A lysine methyltransferase 3","kelch repeat and BTB domain containing 4","sarcosine dehydrogenase","ATP synthase mitochondrial F1 complex assembly factor 1","STIL centriolar assembly protein","Holliday junction recognition protein","collagen type X alpha 1 chain","NADH:ubiquinone oxidoreductase complex assembly factor 4","ubiquitin specific peptidase 45","mortality factor 4 like 2","family with sequence similarity 199, X-linked","RAB9A, member RAS oncogene family","methyltransferase like 8","tetratricopeptide repeat domain 21B","N-myc and STAT interactor","TNF alpha induced protein 6","bromodomain adjacent to zinc finger domain 2B","solute carrier family 36 member 1","lysophosphatidylglycerol acyltransferase 1","basic leucine zipper ATF-like transcription factor 3","G0/G1 switch 2","potassium inwardly rectifying channel subfamily J member 2","RAP2C, member of RAS oncogene family","exosome component 9","phospholipase A2 group XIIA","B9 domain containing 2","coenzyme Q8B","6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2","complement component 4 binding protein alpha","complement component 4 binding protein beta","RAB38, member RAS oncogene family","G protein-coupled receptor 83","argonaute RISC catalytic component 2","MAX dimerization protein 4","CDC28 protein kinase regulatory subunit 2","dynein assembly factor with WD repeats 1","acyl-CoA synthetase long chain family member 3","chondroitin polymerizing factor","aspartyl aminopeptidase","obscurin like cytoskeletal adaptor 1","family with sequence similarity 124 member B","solute carrier family 12 member 4","enkurin domain containing 1","glucosaminyl (N-acetyl) transferase family member 7","CCCTC-binding factor like","family with sequence similarity 210 member B","peptidase inhibitor 3","family with sequence similarity 209 member A","sorting nexin family member 21","secretory leukocyte peptidase inhibitor","WAP four-disulfide core domain 3","alpha tocopherol transfer protein like","phosphatidylinositol-3,4,5-trisphosphate dependent Rac exchange factor 1","potassium voltage-gated channel modifier subfamily S member 1","solute carrier family 12 member 5","Rho GTPase activating protein 40","syndecan 4","nuclear receptor coactivator 3","phosphatidylinositol glycan anchor biosynthesis class T","nuclear receptor coactivator 5","VAMP associated protein B and C","par-6 family cell polarity regulator beta","ATP synthase F1 subunit epsilon","chromodomain helicase DNA binding protein 6","phospholipase C gamma 1","TOX high mobility group box family member 2","serine and arginine rich splicing factor 6","ganglioside induced differentiation associated protein 1 like 1","ADP ribosylation factor guanine nucleotide exchange factor 2","zinc finger NFX1-type containing 1","zinc finger protein 831","chromosome segregation 1 like","TMEM189-UBE2V1 readthrough","RAB22A, member RAS oncogene family","prostaglandin I2 synthase","staufen double-stranded RNA binding protein 1","cadherin 26","snail family transcriptional repressor 1","molybdenum cofactor synthesis 3","syntaxin 16","prostate transmembrane protein, androgen induced 1","ring finger protein 114","DEAD-box helicase 27","chromosome 20 open reading frame 85","breast carcinoma amplified sequence 4","TP53 target 5","Z-DNA binding protein 1","neuralized E3 ubiquitin protein ligase 2","MAGE family member A10","5-methyltetrahydrofolate-homocysteine methyltransferase reductase","FAST kinase domains 3","peptidase D","IQ motif and Sec7 domain ArfGEF 2","vesicle associated membrane protein 7","interleukin 9 receptor","Xg glycoprotein (Xg blood group)","STAM binding protein","N-acetylglucosamine kinase","methylmalonyl-CoA epimerase","poly(A) binding protein interacting protein 2B","small nuclear ribonucleoprotein U4/U6.U5 subunit 27","M-phase phosphoprotein 10","interleukin 17C","ATPase phospholipid transporting 8A1","ubiquitin specific peptidase 22","POF1B actin binding protein","hypoxia inducible factor 3 subunit alpha","zinc finger protein 576","zinc finger protein 45","LY6/PLAUR domain containing 3","CEA cell adhesion molecule 8","ubiquitin specific peptidase 9 X-linked","coagulation factor XIII A chain","glutamate metabotropic receptor 4","transcriptional regulating factor 1","protein kinase C and casein kinase substrate in neurons 1","butyrophilin subfamily 2 member A2","sirtuin 5","magnesium transporter MRS2","WRN helicase interacting protein 1","ribosomal RNA processing 36","small nuclear ribonucleoprotein polypeptide C","serpin family B member 6","exportin 5","ATP binding cassette subfamily C member 10","H1.3 linker histone, cluster member","peroxisomal biogenesis factor 6","N-ribosyldihydronicotinamide:quinone reductase 2",null,"O-acyl-ADP-ribose deacylase 1","unc-5 family C-terminal like","alanyl-tRNA synthetase 2, mitochondrial","H1.1 linker histone, cluster member","zinc finger protein 391","ribosomal protein S10","molybdenum cofactor synthesis 1","H2B clustered histone 11","mediator complex subunit 20","tubulin folding cofactor C","SAM pointed domain containing ETS transcription factor","t-complex 11","MAD2L1 binding protein","apolipoprotein B mRNA editing enzyme catalytic subunit 2","kelch domain containing 3","glycine N-methyltransferase","triggering receptor expressed on myeloid cells 1","male-enhanced antigen 1","kelch like family member 31","collagen type XXI alpha 1 chain","cyclin dependent kinase inhibitor 1A","SRY-box transcription factor 4","glyoxalase I","copine 5","ras responsive element binding protein 1","signal sequence receptor subunit 1","RIO kinase 1","solute carrier family 35 member B3","ribonuclease P/MRP subunit p40","ataxin 1","nucleoporin 153","DEK proto-oncogene","eukaryotic translation elongation factor 1 epsilon 1","RUNX family transcription factor 2","LRR binding FLII interacting protein 1","RAB17, member RAS oncogene family","C-X-C motif chemokine ligand 6","epiregulin","myelin regulatory factor","secretoglobin family 2A member 1","AHNAK nucleoprotein","ER membrane protein complex subunit 3","ssu-2 homolog","SH3 domain and tetratricopeptide repeats 1","CCR4-NOT transcription complex subunit 1","leucine rich repeat containing 29","Bardet-Biedl syndrome 2","metallothionein 1G","metallothionein 2A","chromosome 16 open reading frame 70","glutamic-oxaloacetic transaminase 2","docking protein 4","G protein-coupled receptor 18","citrate lyase beta like","transmembrane O-mannosyltransferase targeting cadherins 4","RAP2A, member of RAS oncogene family","ATP binding cassette subfamily C member 4","ephrin B2","SRY-box transcription factor 21","transmembrane 9 superfamily member 2","homologous recombination factor with OB-fold","kinesin family member 25","interferon regulatory factor 1","UPF3B regulator of nonsense mediated mRNA decay","ring finger protein 113A","septin 6","transmembrane protein 255A","NADH:ubiquinone oxidoreductase subunit A1","distal membrane arm assembly complex 2 like","bone morphogenetic protein 4","prostaglandin E receptor 2","family with sequence similarity 193 member A","G protein-coupled receptor kinase 4","SRY-box transcription factor 9","tektin 3","heparan sulfate-glucosamine 3-sulfotransferase 3B1","solute carrier family 25 member 35","mitochondrial ribosomal protein S7","golgi associated, gamma adaptin ear containing, ARF binding protein 3","armadillo repeat containing 7","nucleoporin 85","solute carrier family 25 member 19","MIF4G domain containing","5', 3'-nucleotidase, cytosolic","misato mitochondrial distribution and morphology regulator 1","chromosome 1 open reading frame 61","transcription termination factor 1","general transcription factor IIIC subunit 4","DEAD-box helicase 31","protein phosphatase 1 regulatory subunit 12C","membrane bound O-acyltransferase domain containing 7","src-related kinase lacking C-terminal regulatory tyrosine and N-terminal myristylation sites","SLC2A4 regulator","fibronectin type III domain containing 11","pancreatic progenitor cell differentiation and proliferation factor","interleukin 1 beta","plasminogen like B2","coiled-coil-helix-coiled-coil-helix domain containing 5","paired box 8","insulin induced gene 2","RNA polymerase I subunit B","coiled-coil domain containing 93","pleckstrin and Sec7 domain containing 4","solute carrier family 25 member 23","persephin","general transcription factor IIF subunit 1","alkB homolog 7","caseinolytic mitochondrial matrix peptidase proteolytic subunit","TNF superfamily member 9","THO complex 2","mediator complex subunit 1","ribosomal protein L23","autophagy related 4C cysteine peptidase","complement C3","SH2 domain containing 3A","thyroid hormone receptor interactor 10","G protein-coupled receptor 108","TNF superfamily member 14","FosB proto-oncogene, AP-1 transcription factor subunit","outer mitochondrial membrane lipid metabolism regulator OPA3","small nuclear ribonucleoprotein D2 polypeptide","reticulon 2","EMAP like 2","vasodilator stimulated phosphoprotein","symplekin","glycerophosphocholine phosphodiesterase 1","syndecan binding protein 2","pantothenate kinase 2","transglutaminase 3","CD93 molecule","GDNF inducible zinc finger protein 1","NSF attachment protein beta","centromere protein B","proteasome inhibitor subunit 1","D-aminoacyl-tRNA deacylase 1","RANBP2-type and C3HC4-type zinc finger containing 1","thioredoxin related transmembrane protein 4","serine/threonine kinase 35","small nuclear ribonucleoprotein polypeptides B and B1","neurensin 2","adaptor related protein complex 5 subunit sigma 1","ribosome binding protein 1","zinc finger protein 133","fibronectin leucine rich transmembrane protein 3","ovo like zinc finger 2","proprotein convertase subtilisin/kexin type 2","McKusick-Kaufman syndrome","beaded filament structural protein 1","destrin, actin depolymerizing factor","small nuclear ribonucleoprotein polypeptide B2","mitochondrial genome maintenance exonuclease 1","TBC1 domain family member 20","inosine triphosphatase","minichromosome maintenance 8 homologous recombination repair factor","transmembrane protein 74B","family with sequence similarity 110 member A","mitochondrial ribosomal protein S26","sphingosine-1-phosphate receptor 4","nicalin","heterogeneous nuclear ribonucleoprotein R","zinc finger protein 436","MYC associated factor X","armadillo repeat containing X-linked 5","matrix metallopeptidase 24","N-terminal EF-hand calcium binding protein 3","inhibitor of DNA binding 1, HLH protein","RALY heterogeneous nuclear ribonucleoprotein","dynein light chain roadblock-type 1","eukaryotic translation initiation factor 2 subunit beta","ERGIC and golgi 3","reactive oxygen species modulator 1","family with sequence similarity 83 member C","BPI fold containing family B member 1","centrosomal protein 250","PLAG1 like zinc finger 2","MMP24 opposite strand","lysine demethylase 5C","angiomotin","transmembrane protein 115","proteasome 20S subunit beta 2","argonaute RISC catalytic component 3","uroporphyrinogen decarboxylase","ST3 beta-galactoside alpha-2,3-sialyltransferase 3","transmembrane protein 53","HECT domain E3 ubiquitin protein ligase 3","kinesin light chain 1","X-ray repair cross complementing 3","tubulin gamma complex associated protein 3","MCF.2 cell line derived transforming sequence like","PCI domain containing 2","leucine rich repeat and fibronectin type III domain containing 3","IGF like family receptor 1","calpain small subunit 1","programmed cell death 2 like","RNA binding motif protein 42","ubiquitin like modifier activating enzyme 2","free fatty acid receptor 2","hematopoietic cell signal transducer","free fatty acid receptor 1","cytochrome c oxidase subunit 6B1","thyroid hormone receptor alpha","C-C motif chemokine receptor 7","nuclear receptor subfamily 1 group D member 1","FERM domain containing 8","peroxiredoxin 5","BCL2 like 12","interferon regulatory factor 3","protein arginine methyltransferase 1","RAS related","proline rich and Gla domain 2","SR-related CTD associated factor 1","proline rich 12","fibronectin leucine rich transmembrane protein 1","argininosuccinate lyase","SBDS ribosome maturation factor","statherin","histatin 1","signal transducer and activator of transcription 5A","WNK lysine deficient protein kinase 4","beclin 1","protein kinase C gamma","TNF receptor associated protein 1","GLIS family zinc finger 2","nuclear speckle splicing regulatory protein 1","DnaJ heat shock protein family (Hsp40) member C8","AT-hook DNA binding motif containing 1","interferon alpha inducible protein 6","zinc finger protein 384","EMG1 N1-specific pseudouridine methyltransferase","ubiquitously expressed prefoldin like chaperone","complement factor properdin","ETS transcription factor ELK1","translocase of inner mitochondrial membrane 17B","pecanex 4","autophagy related 14","kinectin 1","SIX homeobox 1","ras homolog family member J","DLG associated protein 5","trans-L-3-hydroxyproline dehydratase","heat shock protein family A (Hsp70) member 2","zinc finger and BTB domain containing 1","tRNA methyltransferase 5","sphingosine-1-phosphate phosphatase 1","pleckstrin homology and RhoGEF domain containing G3","PZP alpha-2-macroglobulin like","PR/SET domain 7","ras homolog family member T1","ecotropic viral integration site 2A","oligodendrocyte myelin glycoprotein","WD repeat domain 60","allograft inflammatory factor 1 like","family with sequence similarity 78 member A","nucleoporin 214","arginine vasopressin receptor 2","solute carrier family 10 member 3","mitogen-activated protein kinase kinase 2","heterogeneous nuclear ribonucleoprotein H2","armadillo repeat containing X-linked 1","translocase of inner mitochondrial membrane 8A","zinc finger C4H2-type containing","calnexin","integrator complex subunit 11","regulator of G protein signaling 13","inositol-pentakisphosphate 2-kinase","zinc finger protein 484","FYVE, RhoGEF and PH domain containing 3","HIVEP zinc finger 3","phosphopantothenoylcysteine synthetase","BAF chromatin remodeling complex subunit BCL11B","cytochrome c oxidase subunit 7C","TNF receptor associated factor 2","abhydrolase domain containing 8","mannan binding lectin serine peptidase 1","ATPase 13A4","DNA helicase B","RAP1B, member of RAS oncogene family","tetraspanin 8","bestrophin 3","RAB3A interacting protein","protein tyrosine phosphatase receptor type B","dual specificity tyrosine phosphorylation regulated kinase 2","YEATS domain containing 4","taste 2 receptor member 3","taste 2 receptor member 4","taste 2 receptor member 5","leucine rich repeat containing 61","alpha-L-iduronidase","fibroblast growth factor receptor like 1","transmembrane protein 175","aurora kinase A and ninein interacting protein","peptidylprolyl cis/trans isomerase, NIMA-interacting 1","F-box and leucine rich repeat protein 12","ER membrane protein complex subunit 1","ubiquitin protein ligase E3 component n-recognin 4","heterochromatin protein 1 binding protein 3","adhesion G protein-coupled receptor E2","SIN3 transcription regulator family member B","solute carrier family 35 member E1","epidermal growth factor receptor pathway substrate 15 like 1","Kruppel like factor 2","olfactory receptor family 7 subfamily C member 1","F2R like thrombin or trypsin receptor 3","ubiquinol-cytochrome c reductase, complex III subunit XI","growth factor, augmenter of liver regeneration","synaptogyrin 3","protein kinase, membrane associated tyrosine/threonine 1","WAP, follistatin/kazal, immunoglobulin, kunitz and netrin domain containing 1","WD repeat domain 24","F-box and leucine rich repeat protein 16","chromosome transmission fidelity factor 18","microtubule actin crosslinking factor 1","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4","lysine demethylase 4B","toll like receptor adaptor molecule 1","methyltransferase like 25","ER membrane protein complex subunit 6","methyltransferase like 16","tubulin alpha 4a","villin 1","angio associated migratory cell protein","PNKD metallo-beta-lactamase domain containing","TNF receptor superfamily member 19","ring finger protein 6","enoyl-CoA hydratase, short chain 1","zinc finger protein 835","A-kinase anchoring protein 9","SEM1 26S proteasome complex subunit","huntingtin interacting protein 1","protein tyrosine phosphatase non-receptor type 12","cytochrome p450 oxidoreductase","fibrinogen like 2","serine/threonine/tyrosine interacting like 1","STEAP4 metalloreductase","G protein subunit alpha i1","peroxisomal biogenesis factor 1","mitochondrial transcription termination factor 1","sarcoglycan epsilon","RNA binding motif protein 48","CAS1 domain containing 1","zinc finger protein 780B","leucine rich repeat and fibronectin type III domain containing 1","ZFP36 ring finger protein","steroid 5 alpha-reductase 3","phosphoribosylaminoimidazole carboxylase and phosphoribosylaminoimidazolesuccinocarboxamide synthase","phosphoribosyl pyrophosphate amidotransferase","tubulin gamma complex associated protein 6","adrenomedullin 2","DiGeorge syndrome critical region gene 6 like","DGCR8 microprocessor complex subunit","aspartate beta-hydroxylase domain containing 2","stromal cell derived factor 2 like 1","tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein eta","G protein subunit alpha z","beta-1,4-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase","adenosine A2a receptor","activating transcription factor 4","alpha 1,4-galactosyltransferase (P blood group)","CDC42 effector protein 1","apolipoprotein L3","melanin concentrating hormone receptor 1","tyrosylprotein sulfotransferase 2","BAR/IMD domain containing adaptor protein 2 like 2","mercaptopyruvate sulfurtransferase","thiosulfate sulfurtransferase","apolipoprotein L2","Rac family small GTPase 2","LIF interleukin 6 family cytokine","chromosome 22 open reading frame 23","apolipoprotein B mRNA editing enzyme catalytic subunit 3A","apolipoprotein B mRNA editing enzyme catalytic subunit 3F","RIB43A domain with coiled-coils 2","keratin 17","ER membrane protein complex subunit 4","sperm antigen with calponin homology and coiled-coil domains 1","carboxypeptidase A4","dedicator of cytokinesis 4","protection of telomeres 1","ATPase H+ transporting V1 subunit F","LSM8 homolog, U6 small nuclear RNA associated","cadherin related family member 3","PRKR interacting protein 1","podocalyxin like","forkhead box P2","striatin interacting protein 2","intraflagellar transport 22","muskelin 1","DnaJ heat shock protein family (Hsp40) member B9","filamin C","leucine rich repeat containing 4","calumenin","coiled-coil domain containing 136","smoothened, frizzled class receptor","interferon regulatory factor 5","leucine rich repeat containing 17","kelch domain containing 10","NADH:ubiquinone oxidoreductase subunit A5","FEZ family zinc finger 1","mitochondrial ribosomal protein S12","myosin IB","metaxin 2","phosphodiesterase 11A","chimerin 1","glutamate decarboxylase 1","O-sialoglycoprotein endopeptidase like 1","ORMDL sphingolipid biosynthesis regulator 1","histone acetyltransferase 1","HECT and RLD domain containing E3 ubiquitin protein ligase 2","small nuclear ribonucleoprotein polypeptide N","proteasome assembly chaperone 2","twisted gastrulation BMP signaling modulator 1","Rho GTPase activating protein 22","WDFY family member 4","eukaryotic translation initiation factor 2 alpha kinase 4","myosin VC","cingulin like 1","tropomodulin 2","tau tubulin kinase 2","elongation factor for RNA polymerase II 3","coiled-coil domain containing 32","INO80 complex ATPase subunit","interactor of little elongation complex ELL subunit 2","delta like canonical Notch ligand 4","aldehyde dehydrogenase 1 family member A2","MINDY lysine 48 deubiquitinase 2","isovaleryl-CoA dehydrogenase","kinetochore localized astrin (SPAG5) binding protein","deoxyuridine triphosphatase","ChaC glutathione specific gamma-glutamylcyclotransferase 1","CLN6 transmembrane ER protein","cAMP regulated phosphoprotein 19","vacuolar protein sorting 13 homolog C","calmodulin like 4","immunoglobulin superfamily containing leucine rich repeat","THAP domain containing 10","lysyl oxidase like 1","atypical chemokine receptor 4","anaphase promoting complex subunit 13","methyl-CpG binding domain 4, DNA glycosylase","coatomer protein complex subunit beta 1","proteasome 20S subunit alpha 1","sulfatase modifying factor 2","palladin, cytoskeletal associated protein","signal peptidase complex subunit 3","gamma-butyrobetaine hydroxylase 1","secretion regulating guanine nucleotide exchange factor","potassium voltage-gated channel subfamily C member 1","tryptophan hydroxylase 1","cysteine and glycine rich protein 3","E2F transcription factor 8","dCMP deaminase","SRY-box transcription factor 15","PICALM interacting mitotic regulator","RPA interacting protein","ubiquitin specific peptidase 6","phospholipase D2","aryl hydrocarbon receptor interacting protein like 1","CD68 molecule","thioredoxin domain containing 17","ATPase Na+/K+ transporting subunit beta 2","FMR1 autosomal homolog 2","kinesin family member 1C","mannose-P-dolichol utilization defect 1","PHD finger protein 20 like 1","leucine rich repeat containing 6","cyclin T1","pseudouridine synthase 7 like","KRI1 homolog","interleukin enhancer binding factor 3","solute carrier family 44 member 2","adaptor related protein complex 1 subunit mu 2","cyclin dependent kinase inhibitor 2D","microtubule associated scaffold protein 1","sialic acid binding Ig like lectin 9","kallikrein related peptidase 10","neuroguidin","receptor interacting serine/threonine kinase 3","adenylate cyclase 4","RAB2B, member RAS oncogene family","BCL2 like 2","ajuba LIM protein","D-aminoacyl-tRNA deacylase 2","poly(ADP-ribose) polymerase 2","HEAT repeat containing 5A","forkhead box A1","sorting nexin 6","E2F associated phosphoprotein","egl-9 family hypoxia inducible factor 3","MIS18 binding protein 1","neural retina leucine zipper","ribonuclease A family member 1, pancreatic","NEDD8 ubiquitin like modifier","defender against cell death 1","telomerase associated protein 1","erythrocyte membrane protein band 4.1 like 4A","receptor accessory protein 5","integrin alpha FG-GAP repeat containing 1","glutamine rich 2","forkhead box J1","SEC14 like lipid binding 1","rhomboid 5 homolog 2","aralkylamine N-acetyltransferase","Rac/Cdc42 guanine nucleotide exchange factor 6","MAP7 domain containing 3","fibroblast growth factor 13","ASH2 like, histone lysine methyltransferase complex subunit","TELO2 interacting protein 2","ADP-ribosyltransferase 1","cholinergic receptor nicotinic alpha 10 subunit","cyclin dependent kinase inhibitor 1C","shugoshin 1","ribosomal protein S4 Y-linked 1","Kruppel like factor 16","post-glycosylphosphatidylinositol attachment to proteins 6","deoxyhypusine hydroxylase","MAU2 sister chromatid cohesion factor","SHC adaptor protein 2","phospholipid phosphatase related 3","abhydrolase domain containing 17A","synaptotagmin 5","troponin I3, cardiac type","CBFA2/RUNX1 partner transcriptional co-repressor 3","guanidinoacetate N-methyltransferase","pseudouridine 5'-phosphatase","ER membrane associated RNA degradation","PHD finger protein 10","proline rich and Gla domain 3","calcium release activated channel regulator 2A","nucleoredoxin like 2","StAR related lipid transfer domain containing 8","glycerophosphodiester phosphodiesterase domain containing 2","spermidine/spermine N1-acetyltransferase 1","G protein nucleolar 3 like","SH3 domain binding protein 4","motile sperm domain containing 2","dedicator of cytokinesis 6","ECSIT signaling integrator","low density lipoprotein receptor","elongation factor 1 homolog","tetraspanin 16","protein kinase C substrate 80K-H","calponin 1","cell division cycle 16","thioesterase superfamily member 6","nectin cell adhesion molecule 2","apolipoprotein E","translocase of outer mitochondrial membrane 40","apolipoprotein C1","growth arrest and DNA damage inducible gamma","dipeptidyl peptidase like 6","exportin 7","angiotensin I converting enzyme 2","family with sequence similarity 98 member C","scaffold attachment factor B2","ribosomal protein L36","ATPase phospholipid transporting 8B3","kinesin family member 1A","GTP binding protein 3, mitochondrial","plasmalemma vesicle associated protein","bone marrow stromal cell antigen 2","solute carrier family 27 member 1","NOP2/Sun RNA methyltransferase 5","USH1 protein network component harmonin binding protein 1","collagen beta(1-O)galactosyltransferase 1","DET1 and DDB1 associated 1","mitochondrial ribosomal protein L34","6-phosphogluconolactonase","LSM7 homolog, U6 small nuclear RNA and mRNA degradation associated","TUB like protein 4","sorting nexin 9","reticulon 4 interacting protein 1","glutaminyl-tRNA amidotransferase subunit QRSL1","mitochondrial transcription rescue factor 1","radial spoke head 3","MAS1 proto-oncogene, G protein-coupled receptor","MLLT1 super elongation complex subunit","afadin, adherens junction formation factor","actinin alpha 4","serine/threonine kinase 33","NADH:ubiquinone oxidoreductase subunit A10","actin related protein 2/3 complex subunit 1B","calcium voltage-gated channel auxiliary subunit gamma 6","zinc finger SWIM-type containing 6","FCH and mu domain containing endocytic adaptor 1","unc-13 homolog A","microtubule associated protein 1S","kelch domain containing 7B","peroxidasin","single stranded DNA binding protein 4","growth differentiation factor 15","pyroglutamyl-peptidase I","IQ motif containing N","LSM4 homolog, U6 small nuclear RNA and mRNA degradation associated","JunD proto-oncogene, AP-1 transcription factor subunit","histidine rich calcium binding protein","transient receptor potential cation channel subfamily M member 4","zinc finger protein 557","crumbs cell polarity complex component 3","calmodulin regulated spectrin associated protein 1","UBA domain containing 1","S-antigen visual arrestin","zinc finger and BTB domain containing 46","helicase with zinc finger 2","sterile alpha motif domain containing 10","lymphocyte specific protein 1","troponin T3, fast skeletal type","troponin I2, fast skeletal type","collagen type V alpha 1 chain","ataxin 10","tubulin gamma complex associated protein 2","cytochrome P450 family 2 subfamily E member 1","patatin like phospholipase domain containing 7","p21 (RAC1) activated kinase 4","zinc finger protein 337","centrosomal protein 85","TATA-box binding protein associated factor 4","RBBP8 N-terminal like","laminin subunit alpha 5","oxysterol binding protein like 2","adhesion regulating molecule 1","argininosuccinate synthase 1","exosome component 2","protein O-mannosyltransferase 1","uridine-cytidine kinase 1","proline rich coiled-coil 2B","charged multivesicular body protein 2A","ubiquitin conjugating enzyme E2 M","tripartite motif containing 28","methyltransferase like 26","Yip1 domain family member 2","autophagy related 4D cysteine peptidase","eukaryotic translation initiation factor 2 subunit gamma","transmembrane protein 160","zinc finger CCCH-type containing 4","neuronal PAS domain protein 1","glia maturation factor gamma","mitogen-activated protein kinase kinase kinase 10","Rho guanine nucleotide exchange factor 16","leucine rich repeat containing 47","sestrin 2","sphingomyelin phosphodiesterase acid like 3B","ATP synthase inhibitory factor subunit 1","mediator complex subunit 18","thymocyte selection associated family member 2","CAP-Gly domain containing linker protein 1","coiled-coil domain containing 62","huntingtin interacting protein 1 related","zinc finger protein 317","peter pan homolog","eukaryotic translation initiation factor 3 subunit G","shiftless antiviral inhibitor of ribosomal frameshifting","DNA methyltransferase 1","zinc finger protein 426","solute carrier family 6 member 8","pregnancy up-regulated nonubiquitous CaM kinase","dyskerin pseudouridine synthase 1","plexin A3","membrane palmitoylated protein 1","zinc finger protein 331","zinc finger protein 236","LDL receptor related protein 3","chromosome 12 open reading frame 65","nucleolar protein 11","ubiquitination factor E4B","castor zinc finger 1","polycystin family receptor for egg jelly","hydroxysteroid 17-beta dehydrogenase 3","hyaluronan binding protein 4","solute carrier family 35 member D2","proline rich and Gla domain 1","ubiquitin like modifier activating enzyme 1","regucalcin","DNA polymerase nu","peptidylprolyl isomerase like 4","A-kinase anchoring protein 12","spectrin repeat containing nuclear envelope protein 1","UL16 binding protein 3","large tumor suppressor kinase 1","EPS8 like 1","leukocyte immunoglobulin like receptor B2","AAR2 splicing factor","tubulin tyrosine ligase like 9","BPI fold containing family A member 2","RNA binding motif protein 39","zinc finger protein 341","gamma-glutamyltransferase 7","acyl-CoA synthetase short chain family member 2","Cdc42 guanine nucleotide exchange factor 9","glial fibrillary acidic protein","ATPase H+ transporting V1 subunit E1","zinc finger protein 227","zinc finger protein 428","zinc finger protein 141","cytochrome c oxidase subunit 4I1","ER membrane protein complex subunit 8","Gse1 coiled-coil protein","GINS complex subunit 2","charged multivesicular body protein 1A","SH3 domain binding glutamate rich protein like","cytochrome c oxidase subunit 7B","coagulation factor XII","proline rich 7, synaptic","nuclear factor of activated T cells 1","indoleamine 2,3-dioxygenase 1","gap junction protein alpha 9","cyclase associated actin cytoskeleton regulatory protein 1","palmitoyl-protein thioesterase 1","RAB11 family interacting protein 4","ring finger protein, LIM domain interacting","ATP binding cassette subfamily B member 7","TNF receptor associated factor 3","HAUS augmin like complex subunit 8","adhesion G protein-coupled receptor E3","mitochondrial ribosomal protein S25","SH3 domain binding protein 5","2-hydroxyacyl-CoA lyase 1","TBC1 domain family member 5","calpain 7","raftlin, lipid raft linker 1","chromosome 3 open reading frame 20","rabenosyn, RAB effector","polypeptide N-acetylgalactosaminyltransferase 15","solute carrier family 6 member 6","potassium voltage-gated channel subfamily C member 3","napsin A aspartic peptidase","nuclear receptor subfamily 1 group H member 2","PDZ and LIM domain 4","kinesin family member 3A","alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase","glutamine-fructose-6-phosphate transaminase 2","tubulin gamma 1","proteasome activator subunit 3","ribosomal protein L27","PSMC3 interacting protein","amine oxidase copper containing 3","ATP citrate lyase","vacuolar protein sorting 25 homolog","amine oxidase copper containing 2","glucose-6-phosphatase catalytic subunit","NADH:ubiquinone oxidoreductase subunit A2","ankyrin repeat and KH domain containing 1","diaphanous related formin 1","Nedd4 family interacting protein 1","ubiquitin conjugating enzyme E2 D2","exocyst complex component 4","ArfGAP with coiled-coil, ankyrin repeat and PH domains 3","chromosome 1 open reading frame 159","anoctamin 1","PTPRF interacting protein alpha 1","transmembrane protein 204","THO complex 6","TNF receptor associated factor 7","ninjurin 1","carbonic anhydrase 6","nephrocystin 4","microtubule associated protein 1B","Rhox homeobox family member 2","interleukin 13 receptor subunit alpha 1","WD repeat domain 44","creatine kinase, mitochondrial 2","zinc finger CCHC-type containing 9","tensin 4","DNA topoisomerase II alpha","StAR related lipid transfer domain containing 3","retinoic acid receptor alpha","protein phosphatase 1 regulatory inhibitor subunit 1B","KH RNA binding domain containing, signal transduction associated 3","chromodomain helicase DNA binding protein 1 like","peroxisomal biogenesis factor 11 beta","flavin containing dimethylaniline monoxygenase 5","protein inhibitor of activated STAT 3","protein kinase AMP-activated non-catalytic subunit beta 2","pyruvate dehydrogenase E1 alpha 1 subunit","retinoic acid induced 2","methylcrotonoyl-CoA carboxylase 2","zinc finger protein 304","zinc finger and SCAN domain containing 5A","zinc finger protein 132","ubiquitin specific peptidase 29","selenoprotein S","chondroitin sulfate synthase 1","small nuclear ribonucleoprotein polypeptide A'","LLGL scribble cell polarity complex component 1","lin-28 homolog A","THAP domain containing 1","rhophilin Rho GTPase binding protein 2","chromosome 19 open reading frame 12","FA core complex associated protein 24","leucine rich repeat containing 9","actin related protein 10","abhydrolase domain containing 12B","GTP cyclohydrolase 1","galectin 3","podocan like 1","DnaJ heat shock protein family (Hsp40) member B1","zinc finger SWIM-type containing 4","F-box and WD repeat domain containing 9","regulatory factor X1","zinc finger protein 20","chromosome 19 open reading frame 57","DDB1 and CUL4 associated factor 15","coiled-coil and C2 domain containing 1A","tripartite motif containing 21","spermatogenesis associated 6","leucine rich repeat containing 41","chaperonin containing TCP1 subunit 6B","DExH-box helicase 30","Raf-1 proto-oncogene, serine/threonine kinase","peroxisome proliferator activated receptor gamma","nucleoporin 210","Fc receptor like A","hydroxysteroid 17-beta dehydrogenase 7","enolase superfamily member 1","elastin microfibril interfacer 2","SLX1 homolog A, structure-specific endonuclease subunit","ADP ribosylation factor interacting protein 2","tripartite motif containing 5","cyclic nucleotide gated channel subunit alpha 4","tripartite motif containing 22","ribosomal RNA processing 8","translocase of inner mitochondrial membrane 10B","EFR3 homolog A","HERV-H LTR-associating 1","pentatricopeptide repeat domain 3","inner membrane mitochondrial protein","mitochondrial ribosomal protein L35","IQ motif containing with AAA domain 1","ILK associated serine/threonine phosphatase","period circadian regulator 2","receptor activity modifying protein 1","selenocysteine lyase","protein tyrosine phosphatase receptor type E","RAN, member RAS oncogene family","protein kinase AMP-activated catalytic subunit alpha 1","caspase recruitment domain family member 6","RAP1 GTPase activating protein 2","clustered mitochondria homolog","inositol polyphosphate-5-phosphatase K","MYB binding protein 1a","replication protein A1","serpin family F member 1","ubiquitin conjugating enzyme E2 G1","eukaryotic elongation factor, selenocysteine-tRNA specific","TBC1 domain family member 14","transmembrane protein 128","coenzyme Q3, methyltransferase","PNN interacting serine and arginine rich protein","SEC61 translocon gamma subunit","LanC like 2","fidgetin like 1","G-rich RNA sequence binding factor 1","enamelin","joining chain of multimeric IgA and IgM","ankyrin repeat domain 17","UTP3 small subunit processome component","integrin subunit beta 4","WW domain binding protein 2","H3.3 histone B","unk zinc finger","tripartite motif containing 47","zinc finger RANBP2-type containing 2","eukaryotic translation initiation factor 5A","lysine demethylase 6B","C-type lectin domain containing 10A","solute carrier family 52 member 1","G protein pathway suppressor 2","XIAP associated factor 1","discs large MAGUK scaffold protein 4","reactive intermediate imine deaminase A homolog","vacuolar protein sorting 13 homolog B","regulator of G protein signaling 22","matrilin 2","receptor accessory protein 2","pterin-4 alpha-carbinolamine dehydratase 2","stromal cell derived factor 2","flotillin 2","Era like 12S mitochondrial rRNA chaperone 1","protein arginine methyltransferase 7","nucleolar pre-rRNA processing protein NIP7","telomeric repeat binding factor 2","vacuolar protein sorting 4 homolog A","MTSS I-BAR domain containing 2","ankyrin repeat and EF-hand domain containing 1","PC-esterase domain containing 1A","BTB domain containing 3","proliferating cell nuclear antigen","nuclear transport factor 2 like export factor 1","RNA polymerase III subunit F","Ras and Rab interactor 2","protein tyrosine phosphatase receptor type A","death associated protein 3","Rh family B glycoprotein (gene/pseudogene)","KH domain containing 4, pre-mRNA splicing factor","ATPase Na+/K+ transporting subunit alpha 4","brevican","Rho guanine nucleotide exchange factor 11","RAB25, member RAS oncogene family","Fc receptor like 2","DDB1 and CUL4 associated factor 8","synaptotagmin 11","immunoglobulin mu DNA binding protein 2","aminoacylase 3","aldehyde dehydrogenase 3 family member B2","testis expressed metallothionein like protein","metabolism of cobalamin associated C","diphthamide biosynthesis 2","target of EGR1, exonuclease","nuclear autoantigenic sperm protein","mutY DNA glycosylase","catenin beta like 1","lipin 3","zinc finger SWIM-type containing 3","RNA binding motif protein 38","V-set and transmembrane domain containing 2 like","oxidative stress responsive serine rich 1","serine incorporator 3","protein phosphatase 1 regulatory subunit 3D","dimethylglycine dehydrogenase","betaine--homocysteine S-methyltransferase 2","adaptor related protein complex 3 subunit beta 1","zinc finger BED-type containing 3","PATJ crumbs cell polarity complex component","angiopoietin like 3","solute carrier family 14 member 2","F-box protein 44","ciliogenesis and planar polarity effector 2","caspase 9","dynactin subunit 4","phosphodiesterase 6A","ATPase phospholipid transporting 8A2","microtubule associated scaffold protein 2","zinc finger MYM-type containing 5","ubiquitin specific peptidase like 1","exportin 4","transmembrane phosphoinositide 3-phosphatase and tensin homolog 2","proteasome maturation protein","cyclin dependent kinase 8","arachidonate 5-lipoxygenase activating protein","WASP family member 3","cholinergic receptor muscarinic 3","myosin heavy chain 10","phosphatidylethanolamine N-methyltransferase","SCO cytochrome c oxidase assembly protein 1","myosin phosphatase Rho interacting protein","chitinase 3 like 1","phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 beta","dual serine/threonine and tyrosine protein kinase","chitinase 1","solute carrier family 41 member 1","leucine rich repeat containing G protein-coupled receptor 6","transmembrane and coiled-coil domain family 2","doublecortin like kinase 1","cyclin A1","component of oligomeric golgi complex 6","spartin","epithelial stromal interaction 1","periostin","regulatory factor X associated protein","tumor protein, translationally-controlled 1","GPALPP motifs containing 1","stomatin like 3","replication factor C subunit 3","StAR related lipid transfer domain containing 13","MORC family CW-type zinc finger 4","ring finger protein 128","TBC1 domain family member 8B","transcription elongation factor A like 4","family with sequence similarity 104 member A","solute carrier family 39 member 11","EPH receptor B2","serine and arginine repetitive matrix 1","BTB domain containing 2","PML-RARA regulated adaptor molecule 1","lysine methyltransferase 5C","zinc finger protein 414","phosphodiesterase 6B","HSPA (Hsp70) binding protein 1","casein kinase 1 gamma 2","SMC5-SMC6 complex localization factor 1","carnosine dipeptidase 2","mono-ADP ribosylhydrolase 1","WD repeat domain 74","reticulon 3","phospholipase A and acyltransferase 4","phospholipase A and acyltransferase 2","myosin heavy chain 11","FGFR1OP N-terminal like","mediator complex subunit 10","PDZ domain containing 2","MORC family CW-type zinc finger 2","LARGE xylosyl- and glucuronyltransferase 1","glutathione S-transferase theta 2B (gene/pseudogene)","solute carrier family 2 member 11","C1q and TNF related 6","family with sequence similarity 83 member F","SEC14 like lipid binding 4","GTPase, IMAP family member 6","GTPase, IMAP family member 4","aarF domain containing kinase 2","makorin ring finger protein 1","ArfGAP with GTPase domain, ankyrin repeat and PH domain 3","KRAB-A domain containing 1","actin related protein 3B","neurotensin","BTG anti-proliferation factor 1","leucine rich repeats and IQ motif containing 1","chromosome 12 open reading frame 29","ATPase 13A3","DPY30 domain containing 2","transmembrane protein 254","transmembrane O-mannosyltransferase targeting cadherins 1","KRAS proto-oncogene, GTPase","importin 8","leucyl-tRNA synthetase 1","serine peptidase inhibitor, Kazal type 5","inositol monophosphatase 1","leucine rich repeat and coiled-coil centrosomal protein 1","E2F transcription factor 5","carbonic anhydrase 1","coiled-coil domain containing 59","switching B cell complex subunit SWAP70","aryl hydrocarbon receptor nuclear translocator like","lymphatic vessel endothelial hyaluronan receptor 1","adenosine monophosphate deaminase 3","SET binding factor 2","microtubule associated monooxygenase, calponin and LIM domain containing 2","RAS related 2","hydroxysteroid 17-beta dehydrogenase 4","zinc finger C3H1-type containing","store-operated calcium entry associated regulatory factor","ring finger protein 122","double PHD fingers 2","menin 1","ergosterol biosynthesis 28 homolog","D-glutamate cyclase","unc-79 homolog, NALCN channel complex subunit","NUMB endocytic adaptor protein","cation channel sperm associated auxiliary subunit beta","cytochrome c oxidase assembly factor COX16","tetratricopeptide repeat domain 9","mediator complex subunit 6","eukaryotic translation initiation factor 2 subunit alpha","ADAM metallopeptidase domain 20","lysyl oxidase like 2","elongator acetyltransferase complex subunit 3","ADAM like decysin 1","cap binding complex dependent translation initiation factor","maestro","methyl-CpG binding domain protein 2","immediate early response 3 interacting protein 1","mitochondrial ribosomal protein S36","cyclin B1","cyclin dependent kinase 7","CD180 molecule","interleukin 1 receptor associated kinase 2","calcium/calmodulin dependent protein kinase I","THUMP domain containing 3","von Hippel-Lindau tumor suppressor","basic helix-loop-helix family member e40","ADP ribosylation factor like GTPase 8B","ER degradation enhancing alpha-mannosidase like protein 1","cell adhesion molecule L1 like","Meis homeobox 2","diphthamine biosynthesis 6","katanin regulatory subunit B1 like 1","ER membrane protein complex subunit 7","G protein subunit alpha transducin 2","glutathione S-transferase mu 1","pre-mRNA processing factor 38B","tetraspanin 2","glutathione S-transferase mu 3","vav guanine nucleotide exchange factor 3","proline and serine rich coiled-coil 1","3-hydroxy-3-methylglutaryl-CoA synthase 2","protein tyrosine phosphatase non-receptor type 22","sortilin 1","Wnt family member 2B","prostaglandin F2 receptor inhibitor","late endosomal/lysosomal adaptor, MAPK and MTOR activator 5","notch receptor 2","tripartite motif containing 45","choline/ethanolamine phosphotransferase 1","CD101 molecule","V-set domain containing T cell activation inhibitor 1","adaptor related protein complex 4 subunit beta 1","NSF attachment protein gamma","spire type actin nucleation factor 1","periphilin 1","FKBP prolyl isomerase 11","ADP ribosylation factor 3","transmembrane protein 106C","solute carrier family 38 member 2","tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein theta","kinase D interacting substrate 220","grainyhead like transcription factor 1","Rho associated coiled-coil containing protein kinase 2","radical S-adenosyl methionine domain containing 2","lipin 1","cytidine/uridine monophosphate kinase 2","isoamyl acetate hydrolyzing esterase 1 (putative)","lactate dehydrogenase A","serum amyloid A2","anoctamin 3","interleukin 6 signal transducer","follistatin","neuron navigator 1","cell division cycle 73","translocase of inner mitochondrial membrane 17A","crumbs cell polarity complex component 1","endoplasmic reticulum to nucleus signaling 2","ribosomal protein S15a","asparaginyl-tRNA synthetase 1","RAB11 binding and LisH domain, coiled-coil and HEAT repeat containing","F-box DNA helicase 1","RNA binding motif protein 17","interleukin 2 receptor subunit alpha","ankyrin repeat domain 16","enoyl-CoA hydratase domain containing 3","interleukin 15 receptor subunit alpha","cyclin H","histamine receptor H4","transmembrane protein 241","potassium channel tetramerization domain containing 1","Cdk5 and Abl enzyme substrate 1","dedicator of cytokinesis 2","epithelial membrane protein 1","SRY-box transcription factor 5","RAS like estrogen regulated growth inhibitor","killer cell lectin like receptor D1","killer cell lectin like receptor C1","proline rich protein HaeIII subfamily 2","LDL receptor related protein 4","myosin binding protein C, cardiac","damage specific DNA binding protein 2","ubiquitin specific peptidase 26","retrotransposon Gag like 8C","RNA binding motif protein X-linked 2","serine/threonine kinase 26","piwi like RNA-mediated gene silencing 4","pumilio RNA binding family member 1","SPOC domain containing 1","tyrosyl-tRNA synthetase 1","polyhomeotic homolog 2","cell division cycle associated 8","G protein nucleolar 2","argonaute RISC component 4","hook microtubule tethering protein 1","cytochrome P450 family 2 subfamily J member 2","basic transcription factor 3 like 4","terminal uridylyl transferase 4","pre-mRNA processing factor 38A","desmocollin 2","desmoglein 3","ring finger protein 138","elongator acetyltransferase complex subunit 2","desmocollin 3","dystrobrevin alpha","formin homology 2 domain containing 3","tubulin polyglutamylase complex subunit 2","diacylglycerol lipase alpha","solute carrier family 43 member 3","translocase of inner mitochondrial membrane 10","cobalamin binding intrinsic factor","DExH-box helicase 34","fatty acid desaturase 2","transmembrane protein 258","transcobalamin 1","complement component 5a receptor 2","transmembrane protein 165","clock circadian regulator","platelet derived growth factor receptor alpha","gamma-glutamylamine cyclotransferase","collagen type IV alpha 2 chain","claudin 10","DAZ interacting zinc finger protein 1","UBA domain containing 2","arginine and glutamate rich 1","basic, immunoglobulin-like variable motif containing","ERCC excision repair 5, endonuclease","tripeptidyl peptidase 2","protein O-glucosyltransferase 2","cysteinyl-tRNA synthetase 2, mitochondrial","Rho GTPase activating protein 32","STT3 oligosaccharyltransferase complex catalytic subunit A","ADAM metallopeptidase with thrombospondin type 1 motif 8","ETS proto-oncogene 1, transcription factor","solute carrier family 37 member 2","klotho beta","transmembrane p24 trafficking protein 7","APC regulator of WNT signaling pathway","neuronal regeneration related protein","WD repeat domain 36","osteoclast stimulating factor 1","riboflavin kinase","ubiquilin 1","N(alpha)-acetyltransferase 35, NatC auxiliary subunit","chromosome 9 open reading frame 40","annexin A1","cathepsin L","cell migration inducing hyaluronidase 2","ATP/GTP binding protein 1","golgi membrane protein 1","family with sequence similarity 189 member A2","phosphoserine aminotransferase 1","iron-sulfur cluster assembly 1","ADAM metallopeptidase domain 19","hepatitis A virus cellular receptor 2","cyclin J like","TAO kinase 3","ubiquitin specific peptidase 30","serine dehydratase","musashi RNA binding protein 1","F-box protein 21","2'-5'-oligoadenylate synthetase like","harakiri, BCL2 interacting protein","ring finger protein, transmembrane 2","purinergic receptor P2X 4","BICD family like cargo adaptor 1","deltex E3 ubiquitin ligase 1","TRAF-type zinc finger domain containing 1","cyclin D binding myb like transcription factor 1","transmembrane protein 243","coiled-coil domain containing 146","transmembrane protein 60","CD36 molecule","patatin like phospholipase domain containing 8","hypoxia inducible lipid droplet associated","RAD50 interactor 1","SRSF protein kinase 2","kielin cysteine rich BMP regulator","testin LIM domain protein","MyoD family inhibitor domain containing","mitochondrial tRNA translation optimization 1","ankyrin repeat domain 6","KH domain containing 1","centrosomal protein 162","synaptotagmin binding cytoplasmic RNA interacting protein","sorting nexin 14","5'-nucleotidase ecto","melanocortin 2 receptor accessory protein 2","akirin 2","origin recognition complex subunit 3","lebercilin LCA5","mitogen-activated protein kinase kinase kinase 7","proline rich 5 like","LIM domain only 2","PHD finger protein 21A","N-acetyltransferase 10","ETS homologous factor","E74 like ETS transcription factor 5","proline rich and Gla domain 4","cell cycle associated protein 1","ATP synthase membrane subunit c locus 2","DnaJ heat shock protein family (Hsp40) member C14","CD63 molecule","peripherin","advillin","growth differentiation factor 11","glutaminase 2","integrin subunit alpha 7","thymocyte expressed, positive selection associated 1","family with sequence similarity 186 member B","retinol dehydrogenase 5","ArfGAP with GTPase domain, ankyrin repeat and PH domain 2","biogenesis of lysosomal organelles complex 1 subunit 1","cyclin dependent kinase 4","protein phosphatase 1 regulatory inhibitor subunit 1A","trophinin associated protein","tetraspanin 31","transcription factor CP2","coenzyme Q10A","Fas apoptotic inhibitory molecule 2","poly(A) specific ribonuclease subunit PAN2","extra spindle pole bodies like 1, separase","keratin 7","zinc finger CCCH-type containing 10","heterogeneous nuclear ribonucleoprotein A1","activin A receptor type 1B","OS9 endoplasmic reticulum lectin","potassium voltage-gated channel subfamily H member 3","LTV1 ribosome biogenesis factor","microtubule associated protein 7","CD164 molecule","AFG1 like ATPase","NHS like 1","Abelson helper integration site 1","hes related family bHLH transcription factor with YRPW motif 2","cAMP-dependent protein kinase inhibitor beta","sphingomyelin phosphodiesterase 2","microtubule associated monooxygenase, calponin and LIM domain containing 1","RALBP1 associated Eps domain containing 1","syntaxin 11","tec protein tyrosine kinase","protease associated domain containing 1","ssemaphorin 4F","chaperonin containing TCP1 subunit 7","RAB11 family interacting protein 5","SMYD family member 5","dysferlin","coiled-coil domain containing 142","potassium calcium-activated channel subfamily M regulatory beta subunit 4","ubiquitin specific peptidase 15","glucosamine (N-acetyl)-6-sulfatase","carboxypeptidase M","MDM2 proto-oncogene","kelch like family member 36","beta-carotene oxygenase 1","M-phase phosphoprotein 6","carbohydrate sulfotransferase 5","KIAA0513","dynein cytoplasmic 1 light intermediate chain 2","F-box and leucine rich repeat protein 8","formin homology 2 domain containing 1","coiled-coil domain containing 102A","solute carrier family 9 member A5","ZNF670-ZNF695 readthrough (NMD candidate)","pecanex 2","potassium two pore domain channel subfamily K member 1","URB2 ribosome biogenesis homolog","egl-9 family hypoxia inducible factor 1","calpain 9","component of oligomeric golgi complex 2","ATP binding cassette subfamily B member 10","nucleoside-triphosphatase, cancer-related","TATA-box binding protein associated factor 5 like","glutamate-ammonia ligase","syntaxin 6","ribonuclease L","DExH-box helicase 9","KIAA1614","centrosomal protein 350","N-acetylneuraminate pyruvate lyase","niban apoptosis regulator 1","phosphatidylinositol glycan anchor biosynthesis class C","laminin subunit gamma 1","ring finger and CCCH-type domains 1","SP110 nuclear body protein","mitochondrial ribosomal protein L44","paired box 3","dedicator of cytokinesis 10","tubulin tyrosine ligase like 4","ubiquitin specific peptidase 37","5-hydroxytryptamine receptor 2B","integral membrane protein 2C","solute carrier family 19 member 3","serpin family E member 2","DnaJ heat shock protein family (Hsp40) member B2","Wnt family member 10A","transmembrane BAX inhibitor motif containing 1","cytochrome P450 family 27 subfamily A member 1","eukaryotic translation initiation factor 4E family member 2","armadillo repeat containing 9","calcium binding protein 39","cytochrome c oxidase subunit 5B","REV1 DNA directed polymerase","testis specific 10","major facilitator superfamily domain containing 9","transmembrane protein 127","ectodysplasin A receptor","transforming growth factor beta receptor associated protein 1","GRIP and coiled-coil domain containing 2","mitochondrial ribosomal protein S9","chromosome 2 open reading frame 49","ankyrin repeat domain 36","enhancer of polycomb homolog 2","Rho guanine nucleotide exchange factor 4","iron-sulfur cluster assembly enzyme","aldehyde dehydrogenase 1 family member L2","stabilin 2","ubiquitin specific peptidase 44","SCY1 like pseudokinase 2","cytoskeleton associated protein 4","plexin C1","adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 2","PWP1 homolog, endonuclein","DNA damage regulated autophagy modulator 1","WASH complex subunit 4","solute carrier family 41 member 2","villin like","filamin B","NIMA related kinase 3","vacuolar protein sorting 36 homolog","ribonuclease H2 subunit B","cytoskeleton associated protein 2","TBC1 domain family member 4","thrombospondin type 1 domain containing 1","BORA aurora kinase A activator","leucine rich repeats and calponin homology domain containing 1","succinate-CoA ligase ADP-forming beta subunit","RCC1 and BTB domain containing protein 1","mediator complex subunit 4","PHD finger protein 11","component of oligomeric golgi complex 3","LIM domain 7","sciellin","integral membrane protein 2B","sprouty RTK signaling antagonist 2","nudix hydrolase 15","endothelin receptor type B","RCC1 and BTB domain containing protein 2","lymphocyte cytosolic protein 1","SET domain bifurcated histone lysine methyltransferase 2","secernin 1","chromosome 7 open reading frame 25","tensin 3","speedy/RINGO cell cycle regulator family member E1","carbohydrate sulfotransferase 12","insulin like growth factor 2 mRNA binding protein 3","glycoprotein nmb","Rap guanine nucleotide exchange factor 5","Rac family small GTPase 1","KDEL endoplasmic reticulum protein retention receptor 2","nucleoporin 42","interleukin 6","zinc finger DHHC-type containing 4","acyloxyacyl hydrolase","basic leucine zipper and W2 domains 2","diacylglycerol kinase beta","transforming growth factor beta regulator 4","DEAD-box helicase 56","HUS1 checkpoint clamp component","NAC alpha domain containing","drebrin like","CCM2 scaffold protein","myosin IG","tweety family member 3","cell death inducing DFFA like effector b","tetratricopeptide repeat domain 5","zinc finger homeobox 2","methenyltetrahydrofolate synthetase","ADAM metallopeptidase with thrombospondin type 1 motif 7","abhydrolase domain containing 17C","iron responsive element binding protein 2","alpha kinase 3","transmembrane 6 superfamily member 1","calcium and integrin binding family member 2","calcium binding and coiled-coil domain 2","radical S-adenosyl methionine domain containing 1","N-myristoyltransferase 1","MYCBP associated protein","serine and arginine rich splicing factor 1","vascular endothelial zinc finger 1","chondroadherin","translational activator of cytochrome c oxidase I","testis expressed 2","DDB1 and CUL4 associated factor 7","LIM domain containing 2","BRCA1 interacting protein C-terminal helicase 1","lysine acetyltransferase 7","receptor transporter protein 4","actin like 6A","NADH:ubiquinone oxidoreductase subunit B5","mitochondrial ribosomal protein L47","transformer 2 beta homolog","sodium voltage-gated channel alpha subunit 2","T-box brain transcription factor 1","membrane associated ring-CH-type finger 7","ermin","polypeptide N-acetylgalactosaminyltransferase 5","sodium voltage-gated channel alpha subunit 7","TRAF family member associated NFKB activator","BLK proto-oncogene, Src family tyrosine kinase","SKI like proto-oncogene","glutamyl-prolyl-tRNA synthetase 1","H2.0 like homeobox","vacuolar protein sorting 45 homolog","interleukin 10","potassium channel tetramerization domain containing 3","ribosomal protein S6 kinase C1","COBW domain containing 2","interleukin 1 receptor antagonist","interleukin 36 alpha","interleukin 36 receptor antagonist","sphingomyelin phosphodiesterase 4","WD repeat domain 33","coiled-coil domain containing 115","Sin3A associated protein 130","bridging integrator 1","IMP U3 small nucleolar ribonucleoprotein 4","heparan sulfate 6-O-sulfotransferase 1","UDP-glucose glycoprotein glucosyltransferase 1","glycophorin C (Gerbich blood group)","signal transducing adaptor molecule","glutamate decarboxylase 2","abl interactor 1","YME1 like 1 ATPase","DnaJ heat shock protein family (Hsp40) member C1","nipsnap homolog 3A","leucine rich repeat containing 8 VRAC subunit A","cyclin dependent kinase 9","thioredoxin","outer dense fiber of sperm tails 2","Ecm29 proteasome adaptor and scaffold","torsin family 1 member B","chromosome 9 open reading frame 78","structural maintenance of chromosomes 2","Kruppel like factor 4","torsin family 1 member A","Ral GEF with PH domain and SH3 binding motif 1","niban apoptosis regulator 2","ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 4","DAB2 interacting protein","syntaxin binding protein 1","solute carrier family 2 member 8","angiopoietin like 2","CDK5 regulatory subunit associated protein 2","ZFP37 zinc finger protein","solute carrier family 31 member 2","solute carrier family 31 member 1","toll like receptor 4","zinc finger protein 189","aldolase, fructose-bisphosphate B","syntaxin 17","pre-mRNA processing factor 4","folylpolyglutamate synthase","ubiquitin specific peptidase 20","bile acid-CoA:amino acid N-acyltransferase","ATPase H+ transporting V1 subunit G1","testis expressed 10","GTPase activating Rap/RanGAP domain like 3","mitochondrial ribosomal protein L50","dolichyl-phosphate mannosyltransferase subunit 2, regulatory","WD repeat domain 38","thiosulfate sulfurtransferase like domain containing 2","gamma-aminobutyric acid type B receptor subunit 2","hemogen","proteasome 20S subunit beta 7","tRNA methyltransferase O","Rab9 effector protein with kelch motifs","golgin A1","XPA, DNA damage recognition and repair factor","nuclear cap binding protein subunit 1","acidic nuclear phosphoprotein 32 family member B","phosducin like","ribosomal protein L35","cathepsin V","LIM homeobox transcription factor 1 beta","actin related protein 2/3 complex subunit 5 like","ectonucleotide pyrophosphatase/phosphodiesterase 2","DNA replication and sister chromatid cohesion 1","derlin 1","MYC proto-oncogene, bHLH transcription factor","interleukin 33","distal membrane arm assembly complex 1","RAN binding protein 6","RNA polymerase I subunit E","phospholipase A2 activating protein","interleukin 11 receptor subunit alpha","ubiquitin associated protein 2","aprataxin","ring finger protein 38","talin 1","signaling threshold regulating transmembrane adaptor 1","doublesex and mab-3 related transcription factor 1","DnaJ heat shock protein family (Hsp40) member B5","sperm associated antigen 8","dynactin subunit 3","CD72 molecule","transmembrane protein 8B","glyoxylate and hydroxypyruvate reductase","aldehyde dehydrogenase 1 family member B1","histidine triad nucleotide binding protein 2","Rho guanine nucleotide exchange factor 39","insulin like growth factor binding protein like 1","DENN domain containing 4C","ribosomal protein S6","canopy FGF signaling regulator 3","forkhead box P4","peptidylprolyl isomerase like 1","kinesin light chain 4","kinesin family member 13A","zinc finger and SCAN domain containing 9","Pim-1 proto-oncogene, serine/threonine kinase","guanosine monophosphate reductase","cap methyltransferase 1","transcription factor AP-2 alpha","Yip1 domain family member 3","transmembrane protein 14B","transmembrane protein 63B","fibroblast growth factor receptor substrate 3","tight junction associated protein 1","KIAA0319","interferon regulatory factor 4","solute carrier family 22 member 23","tubulin beta 2A class IIa","leucine rich repeat containing 1","glial cells missing transcription factor 1","biphenyl hydrolase like","receptor interacting serine/threonine kinase 1","ubiquinol-cytochrome c reductase complex assembly factor 2","high mobility group AT-hook 1","transcription factor 19","flotillin 1","immediate early response 3","mediator of DNA damage checkpoint 1","piggyBac transposable element derived 1","alpha tubulin acetyltransferase 1","thiopurine S-methyltransferase","ring finger protein 144B","nurim","mitochondrial carrier 1","valyl-tRNA synthetase 2, mitochondrial","TATA-box binding protein associated factor 8","family with sequence similarity 8 member A1","chromosome 6 open reading frame 52","fibroblast growth factor binding protein 1","fibroblast growth factor binding protein 2","cytoplasmic polyadenylation element binding protein 2","FH2 domain containing 1","toll like receptor 2","tetratricopeptide repeat domain 29","myosin VIIA","FCH and double SH3 domains 2","arrestin beta 1","solute carrier organic anion transporter family member 2B1","THAP domain containing 12","ankyrin repeat domain 42","interleukin 18 binding protein","nuclear mitotic apparatus protein 1","coiled-coil domain containing 90B","synaptotagmin like 2","RAB30, member RAS oncogene family","CREB/ATF bZIP transcription factor","prolylcarboxypeptidase","asparaginyl-tRNA synthetase 2, mitochondrial","ring finger protein 121","mitochondrial ribosomal protein L15","alpha tocopherol transfer protein","gamma-glutamyl hydrolase","solute carrier organic anion transporter family member 5A1","sulfatase 1","trimethylguanosine synthase 1","syndecan binding protein","NIMA related kinase 1","DExD/H-box helicase 60","sortilin related receptor 1","transmembrane serine protease 4","BUD13 homolog","transient receptor potential cation channel subfamily C member 6","matrix metallopeptidase 7","cilia and flagella associated protein 300","defective in cullin neddylation 1 domain containing 5","Yes associated protein 1","tripartite motif containing 29","solute carrier family 37 member 4","BTG anti-proliferation factor 4","POU class 2 homeobox 3","radixin","protein phosphatase 2 scaffold subunit Abeta","ferredoxin 1","chromosome 11 open reading frame 1","FXYD domain containing ion transport regulator 6","Rho GTPase activating protein 20","matrix metallopeptidase 13","transmembrane serine protease 13","caspase 1","caspase 5","alkB homolog 8, tRNA methyltransferase","mitogen-activated protein kinase kinase 5","unc-13 homolog C","sulfide quinone oxidoreductase","CTD small phosphatase like 2","SAFB like transcription modulator","thrombospondin 1","mitogen-activated protein kinase binding protein 1","nucleolar and spindle associated protein 1","NADH:ubiquinone oxidoreductase complex assembly factor 1","kinesin family member 23","integrin subunit alpha 11","kinetochore scaffold 1","HAUS augmin like complex subunit 2","RTF1 homolog, Paf1/RNA polymerase II complex component","poly(ADP-ribose) polymerase family member 6","ribosomal protein lateral stalk subunit P1","progestin and adipoQ receptor family member 5","leucine rich repeat containing 49","tubulin gamma complex associated protein 4","regulator of microtubule dynamics 3","inositol-trisphosphate 3-kinase A","uveal autoantigen with coiled-coil domains and ankyrin repeats","SMAD family member 6","phospholipase C beta 2","transmembrane protein 62","p21 (RAC1) activated kinase 6","ADAM metallopeptidase domain 10","dual oxidase 1","solute carrier family 28 member 2","stimulated by retinoic acid 6","cytochrome P450 family 19 subfamily A member 1","zinc finger protein 280D","semaphorin 6D","ribosomal L24 domain containing 1","spectrin beta, non-erythrocytic 5","GRINL1A complex locus 1","GTP cyclohydrolase I feedback regulator","BCAR3 adaptor protein, NSP family member","tubulin tyrosine ligase like 7","formin binding protein 1 like","kynurenine aminotransferase 3","general transcription factor IIB","Rab geranylgeranyltransferase subunit beta","interferon induced protein 44 like","GIPC PDZ domain containing family member 2","Rho GTPase activating protein 29","interferon induced protein 44","solute carrier family 44 member 5","chloride channel accessory 2","dihydrolipoamide branched chain transacylase E2","RNA 3'-terminal phosphate cyclase","intraflagellar transport 172","selenoprotein I","hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta","adenylate cyclase 3","protein phosphatase, Mg2+/Mn2+ dependent 1B","polyribonucleotide nucleotidyltransferase 1","dynein cytoplasmic 2 light intermediate chain 1","THUMP domain containing 2","cytochrome P450 family 1 subfamily B member 1","RAB1A, member RAS oncogene family","actin related protein 2","prolactin regulatory element binding","solute carrier family 5 member 6","ATP binding cassette subfamily G member 5","prolyl endopeptidase like","solute carrier family 3 member 1","F-box protein 11","SIX homeobox 3","all-trans retinoic acid induced differentiation factor","centromere protein O","leucine rich pentatricopeptide repeat containing","dystrobrevin beta","actin related protein 1A","cytochrome P450 family 2 subfamily C member 9","major facilitator superfamily domain containing 13A","cytochrome P450 family 2 subfamily C member 8","myoferlin","lysyl oxidase like 4","STAM binding protein like 1","cholesterol 25-hydroxylase","ATPase family AAA domain containing 1","BTB domain containing 16","kinesin family member 11","transforming acidic coiled-coil containing protein 2","dual specificity phosphatase 5","calcium homeostasis modulator family member 2","ADP ribosylation factor like GTPase 3","centrosomal protein 55","kinesin family member 20B","ectonucleoside triphosphate diphosphohydrolase 1","exocyst complex component 6","phospholipase C epsilon 1","debranching RNA lariats 1","DnaJ heat shock protein family (Hsp40) member C13","G protein-coupled receptor 87","annexin A7","family with sequence similarity 149 member B1","activating signal cointegrator 1 complex subunit 1","zinc finger protein 365","oncoprotein induced transcript 3","ADAM metallopeptidase with thrombospondin type 1 motif 14","ribosomal protein S24","tet methylcytosine dioxygenase 1","DNA replication helicase/nuclease 2","myopalladin","aldehyde oxidase 1","5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a like 1","BRCA1 associated RING domain 1","signal transducer and activator of transcription 4","calcium responsive transcription factor","asparagine synthetase domain containing 1","methyltransferase like 5","small RNA binding exonuclease protection factor La","NGFI-A binding protein 1","peptidylprolyl isomerase G","FAST kinase domains 1","malate dehydrogenase 1B","HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2","isocitrate dehydrogenase (NADP(+)) 1","Obg like ATPase 1","corepressor interacting with RBPJ, 1","ITPR interacting domain containing 2","cholinergic receptor nicotinic alpha 1 subunit","family with sequence similarity 117 member B","WD repeat domain 12","abl interactor 2","integrin subunit alpha V","solute carrier family 40 member 1","solute carrier family 35 member A5","solute carrier family 49 member 4","SUMO specific peptidase 7","cytochrome c oxidase copper chaperone COX17","poly(ADP-ribose) polymerase family member 9","meiosis specific nuclear structural 1","ubiquitin specific peptidase 8","SECIS binding protein 2 like","tropomodulin 3","signal peptide peptidase like 2A","glucuronic acid epimerase","Src homology 2 domain containing F","aph-1 homolog B, gamma-secretase subunit","integrator complex subunit 14","poly(ADP-ribose) polymerase family member 16","phosphopantothenoylcysteine decarboxylase","hyperpolarization activated cyclic nucleotide gated potassium channel 4","semaphorin 7A (John Milton Hagen blood group)","ubiquitin like 7","Rho GTPase activating protein 24","family with sequence similarity 13 member A","HECT and RLD domain containing E3 ubiquitin protein ligase 3","HECT and RLD domain containing E3 ubiquitin protein ligase family member 6","HECT and RLD domain containing E3 ubiquitin protein ligase 5","zinc finger GRF-type containing 1","adaptor related protein complex 1 associated regulatory protein","COP9 signalosome subunit 4","heterogeneous nuclear ribonucleoprotein D","protein kinase cGMP-dependent 2","RasGEF domain family member 1B","SEC31 homolog A, COPII coat complex component","fibroblast growth factor 5","glycerol-3-phosphate acyltransferase 3","fibroblast growth factor 2","Bardet-Biedl syndrome 7","KIAA1109","bone morphogenetic protein receptor type 1B","Rap1 GTPase-GDP dissociation stimulator 1","La ribonucleoprotein 1B","phosphodiesterase 5A","PR/SET domain 5","transient receptor potential cation channel subfamily C member 3","N-acylethanolamine acid amidase","nucleoporin 54","C-X-C motif chemokine ligand 9","BMP2 inducible kinase","G3BP stress granule assembly factor 2","septin 11","Fraser extracellular matrix complex subunit 1","scavenger receptor class B member 2","cyclin G2","CCR4-NOT transcription complex subunit 6 like","USO1 vesicle transport factor","cyclin dependent kinase like 2","shroom family member 3","annexin A3","inorganic pyrophosphatase 2","centromere protein E","glutathione S-transferase C-terminal domain containing","integrator complex subunit 12","caspase 6","lymphoid enhancer binding factor 1","hydroxyacyl-CoA dehydrogenase","epidermal growth factor","3'-phosphoadenosine 5'-phosphosulfate synthase 1","SEC24 homolog B, COPII coat complex component","chromosome 4 open reading frame 17","protein phosphatase 3 catalytic subunit alpha","solute carrier family 39 member 8","microsomal triglyceride transfer protein","fibrillin 2","mitogen-activated protein kinase 8 interacting protein 3","regulator of G protein signaling 3","guanylyl cyclase domain containing 1","ring finger protein 185","parvin gamma","beta-1,4-N-acetyl-galactosaminyltransferase 3","endoplasmic reticulum protein 27","ETS variant transcription factor 6","GABA type A receptor associated protein like 1","kinesin family member 21A","copine 8","tyrosyl-tRNA synthetase 2","FYVE, RhoGEF and PH domain containing 4","ALG10 alpha-1,2-glucosyltransferase","phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 gamma","SIN3-HDAC complex associated factor","AE binding protein 2","electron transfer flavoprotein subunit beta lysine methyltransferase","ethanolamine kinase 1","zinc finger CCHC-type and RNA binding motif containing 1","transmembrane protein 117","prickle planar cell polarity protein 1","complement C1r subcomponent like","NADH:ubiquinone oxidoreductase subunit A9","calsyntenin 3","killer cell lectin like receptor G1","vesicle associated membrane protein 1","TAP binding protein like","CD27 molecule","retinol binding protein 5","peroxisomal biogenesis factor 5","solute carrier family 38 member 4","adhesion molecule with Ig like domain 2","SR-related CTD associated factor 11","LLP homolog, long-term synaptic facilitation factor","leucine rich repeats and immunoglobulin like domains 3","membrane associated ring-CH-type finger 9","inhibin subunit beta E","GLI pathogenesis related 1","tryptophan hydroxylase 2","pleckstrin homology like domain family A member 1","transmembrane protein 19","protein tyrosine phosphatase receptor type Q","dual specificity phosphatase 6","POC1 centriolar protein B","transmembrane O-mannosyltransferase targeting cadherins 3","small nuclear ribonucleoprotein polypeptide F","amidohydrolase domain containing 1","NEDD1 gamma-tubulin ring complex targeting factor","synaptonemal complex protein 3","growth arrest specific 2 like 3","transmembrane protein 132B","solute carrier family 15 member 4","thymine DNA glycosylase","RBPJ interacting and tubulin associated 1","serine dehydratase like","metabolism of cobalamin associated B","glycolipid transfer protein","GIT ArfGAP 2","trichoplein keratin filament binding","family with sequence similarity 222 member A","forkhead box N4","nucleoporin 58","myotubularin related protein 6","solute carrier family 46 member 3","solute carrier family 7 member 1","ligand of numb-protein X 2","sulfite oxidase","coiled-coil domain containing 65","TARBP2 subunit of RISC loading complex","retinol dehydrogenase 16","desert hedgehog signaling molecule","activin A receptor like type 1","G protein-coupled receptor 84","nucleic acid binding protein 2","NEDD4 binding protein 2 like 1","SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily c member 2","BRCA2 DNA repair associated","KAT8 regulatory NSL complex subunit 2","ceramide synthase 5","mitogen-activated protein kinase kinase kinase 12","integrin subunit beta 7","polypeptide N-acetylgalactosaminyltransferase 6","cysteine sulfinic acid decarboxylase","limb development membrane protein 1 like","chromosome 12 open reading frame 10","extended synaptotagmin 1","transmembrane BAX inhibitor motif containing 6","ankyrin repeat domain 52","zinc finger protein 740","WD repeat and FYVE domain containing 2","heterogeneous nuclear ribonucleoprotein A1 like 2","lysophosphatidic acid receptor 6","esterase D","RB transcriptional corepressor 1","strawberry notch homolog 1","MORN repeat containing 3","SET domain containing 1B, histone lysine methyltransferase","VPS33A core subunit of CORVET and HOPS complexes","VPS37B subunit of ESCRT-I","ras homolog family member F, filopodia associated","density regulated re-initiation and release factor","diaphanous related formin 3","SLAIN motif family member 1","RNA binding motif protein 26","serine/arginine repetitive matrix 4","muscleblind like splicing regulator 2","ring finger protein 113B","abhydrolase domain containing 13","RAB20, member RAS oncogene family","growth hormone regulated TBC protein 1","cullin 4A","tetratricopeptide repeat domain 6","cadherin 24","RRAD and GEM like GTPase 2","cerebellin 3 precursor","testis specific serine kinase 4","NOVA alternative splicing regulator 1","thioredoxin related transmembrane protein 1","FERM domain containing 6","pellino E3 ubiquitin protein ligase family member 2","reticulon 1","armadillo like helical domain containing 4","synaptotagmin 16","solute carrier family 38 member 6","N(alpha)-acetyltransferase 30, NatC catalytic subunit","ADAM metallopeptidase domain 21","retinol dehydrogenase 12","DDB1 and CUL4 associated factor 5","RAB15, member RAS oncogene family","WD repeat domain 89","estrogen receptor 2","stonin 2","EF-hand calcium binding domain 11","G protein-coupled receptor 65","prostaglandin reductase 2","Jun dimerization protein 2","adenylate kinase 7","family with sequence similarity 181 member A","solute carrier family 24 member 4","fibulin 5","serpin family A member 10","clathrin binding box of aftiphilin containing 1","tryptophanyl-tRNA synthetase 1","solute carrier family 25 member 47","WD repeat domain 20","NIPA magnesium transporter 2","solute carrier family 12 member 6","dual oxidase maturation factor 1","microfibril associated protein 1","transcription factor 12","sorbitol dehydrogenase","small EDRK-rich factor 2","zinc finger and SCAN domain containing 29","dual oxidase maturation factor 2","dual oxidase 2","LysM domain containing 2","solute carrier family 27 member 2","fibroblast growth factor 7","histidine decarboxylase","glucosaminyl (N-acetyl) transferase 3, mucin type","BCL2 interacting protein 2","general transcription factor IIA subunit 2","signal recognition particle 14","bromo adjacent homology domain containing 1","dispatched RND transporter family member 2","codanin 1","TLE family member 3, transcriptional corepressor","acidic nuclear phosphoprotein 32 family member A","COMM domain containing 4","ubiquitin conjugating enzyme E2 Q2","proline-serine-threonine phosphatase interacting protein 1","electron transfer flavoprotein subunit alpha","BCL2 related protein A1","high mobility group 20A","S-phase cyclin A associated protein in the ER","tetraspanin 3","WD repeat domain 61","nuclear receptor coactivator 2","nei like DNA glycosylase 1","mannosidase alpha class 2C member 1","DnaJ heat shock protein family (Hsp40) member A4","talin rod domain containing 1","tropomyosin 1","insulin like growth factor 1 receptor","arrestin domain containing 4","PIF1 5'-to-3' DNA helicase","ubiquitin specific peptidase 3","cytochrome P450 family 11 subfamily A member 1","Bardet-Biedl syndrome 4","promyelocytic leukemia","ADAM metallopeptidase with thrombospondin type 1 motif 17","lines homolog 1","unc-51 like kinase 3","proprotein convertase subtilisin/kexin type 6","coiled-coil domain containing 33","CUGBP Elav-like family member 6","secretory carrier membrane protein 2","cytochrome P450 family 1 subfamily A member 2","lectin, mannose binding 1 like","hyaluronan and proteoglycan link protein 3","Rh family C glycoprotein","DNA polymerase gamma, catalytic subunit","retinaldehyde binding protein 1","FA complementation group I","abhydrolase domain containing 2","WD repeat domain 93","TOPBP1 interacting checkpoint and replication regulator","neurotrophic receptor tyrosine kinase 3","DET1 partner of COP1 E3 ubiquitin ligase","milk fat globule-EGF factor 8 protein","zinc finger protein 710","unc-45 myosin chaperone A","multiple C2 and transmembrane domain containing 2","furin, paired basic amino acid cleaving enzyme","IQ motif containing GTPase activating protein 1","CREB regulated transcription coactivator 3","elongation factor like GTPase 1","SH3 domain containing GRB2 like 3, endophilin A3","SEC11 homolog A, signal peptidase complex subunit","glyoxylate reductase 1 homolog","phosphomannomutase 2","solute carrier family 5 member 2","integrin subunit alpha X","transforming growth factor beta 1 induced transcript 1","chromosome 16 open reading frame 58","armadillo repeat containing 5","poly(A)-specific ribonuclease","FTO alpha-ketoglutarate dependent dioxygenase","ubiquinol-cytochrome c reductase core protein 2","cerebellar degeneration related protein 2","immunoglobulin superfamily member 6","Rho GTPase activating protein 17","myosin light chain kinase 3","NKD inhibitor of WNT signaling pathway 1","DEAH-box helicase 38","thioredoxin like 4B","MARVEL domain containing 3","carbohydrate sulfotransferase 4","zinc finger homeobox 3","C-type lectin domain family 18 member B","copine 2","NLR family CARD domain containing 5","katanin regulatory subunit B1","kinesin family member C3","nudix hydrolase 7","glycine cleavage system protein H","CKLF like MARVEL transmembrane domain containing 3","CKLF like MARVEL transmembrane domain containing 2","cadherin 11","nucleolar protein 3","microtubule associated protein 1 light chain 3 beta","membrane bound transcription factor peptidase, site 1","cadherin 13","zinc finger CCHC-type containing 14","MTOR associated protein, eak-7 homolog","adenosine deaminase domain containing 2","oxidative stress induced growth inhibitor 1","interferon regulatory factor 8","ras homolog family member T2","ribosomal protein L3 like","zinc finger and SCAN domain containing 32","ribosomal protein S2","NADH:ubiquinone oxidoreductase subunit B10","3-phosphoinositide dependent protein kinase 1","tigger transposable element derived 7","differentially expressed in FDCP 8 homolog","transcription factor 25","galactosamine (N-acetyl)-6-sulfatase","growth arrest specific 8","mediator complex subunit 9","nuclear receptor corepressor 1","COP9 signalosome subunit 3","GID complex subunit 4 homolog","zinc finger protein 287","kinase suppressor of ras 1","UTP4 small subunit processome component","RAN binding protein 10","chymotrypsin like","glucose-fructose oxidoreductase domain containing 2","NIN1 (RPN12) binding protein 1 homolog","phosphoribosyl pyrophosphate synthetase associated protein 2","unc-45 myosin chaperone B","phosphatidylcholine transfer protein","olfactory receptor family 4 subfamily D member 1","target of myb1 like 1 membrane trafficking protein","chromosome 17 open reading frame 80","transducer of ERBB2, 1","VPS53 subunit of GARP complex","small G protein signaling modulator 2","aminopeptidase puromycin sensitive","src kinase associated phosphoprotein 1","leucine rich repeat containing 46","secernin 2","slingshot protein phosphatase 2","arylsulfatase G","ATP binding cassette subfamily A member 8","glucose-6-phosphatase catalytic subunit 3","clathrin heavy chain","BCAS3 microtubule associated cell migration factor","peptidyl-tRNA hydrolase 2","SS18 subunit of BAF chromatin remodeling complex","TATA-box binding protein associated factor 4b","AFG3 like matrix AAA peptidase subunit 2","PRELI domain containing 3A","inositol monophosphatase 2","G protein subunit alpha L","solute carrier family 39 member 6","regulation of nuclear pre-mRNA domain containing 1A","chromosome 18 open reading frame 21","polypeptide N-acetylgalactosaminyltransferase 1","ASXL transcriptional regulator 3","meprin A subunit beta","GRB2 associated regulator of MAPK1 subtype 1","establishment of sister chromatid cohesion N-acetyltransferase 1","oxysterol binding protein like 1A","GATA binding protein 6","GREB1 like retinoic acid receptor coactivator","regulator of MON1-CCZ1","proline, glutamate and leucine rich protein 1","NPC intracellular cholesterol transporter 1","solute carrier family 14 member 1 (Kidd blood group)","arrestin beta 2","solute carrier family 13 member 5","zinc finger MYND-type containing 15","WD repeat containing antisense to TP53","misshapen like kinase 1","spermidine/spermine N1-acetyltransferase family member 2","asialoglycoprotein receptor 1","phosphoinositide-3-kinase regulatory subunit 5","tumor protein p53","coiled-coil domain containing 40","Rho GDP dissociation inhibitor alpha","transmembrane channel like 6","solute carrier family 16 member 3","caspase recruitment domain family member 14","tweety family member 2","RAB40B, member RAS oncogene family","eukaryotic translation initiation factor 4A3","casein kinase 1 delta","anaphase promoting complex subunit 11","tubulin folding cofactor D","fructosamine 3 kinase related protein","nuclear prelamin A recognition factor","regulatory associated protein of MTOR complex 1","forkhead box K2","tripartite motif containing 65","chromobox 8","secreted and transmembrane 1","ring finger protein 157","centrosomal protein 131","zinc finger protein 750","WD repeat domain 45B","chromobox 4","ring finger protein 165","dymeclin","elaC ribonuclease Z 1","methyl-CpG binding domain protein 1","SMAD family member 4","TNF receptor superfamily member 11a","zinc finger CCHC-type containing 2","F-box protein 15","cerebellin 2 precursor","phorbol-12-myristate-13-acetate-induced protein 1","prolyl 3-hydroxylase family member 4 (inactive)","5'-nucleotidase, cytosolic IIIB","reticulophagy regulator family member 3","erb-b2 receptor tyrosine kinase 2","growth factor receptor bound protein 7","migration and invasion enhancer 1","SH3 and cysteine rich domain 2","insulin like growth factor binding protein 4","FKBP prolyl isomerase 10","thioredoxin like 4A","calcium voltage-gated channel subunit alpha1 A","MISP family member 3","sterile alpha motif domain containing 1","bromodomain containing 4","solute carrier family 39 member 3","nuclear factor I C","tubulin polyglutamylase complex subunit 1","phospholipid phosphatase 2","PR/SET domain 15","phosphofructokinase, liver type","fem-1 homolog A","vav guanine nucleotide exchange factor 1","multivesicular body subunit 12A","SH3 domain containing GRB2 like 1, endophilin A2","dihydrouridine synthase 3 like","dipeptidyl peptidase 9","coiled-coil domain containing 97","transmembrane protein 91","ZFP14 zinc finger protein","sirtuin 3","interferon induced transmembrane protein 3","protein-glucosylgalactosylhydroxylysine glucosidase","hormonally up-regulated Neu-associated kinase","collagen type VI alpha 1 chain","interferon alpha and beta receptor subunit 1","superoxide dismutase 1","collagen type VI alpha 2 chain","salt inducible kinase 1","transient receptor potential cation channel subfamily M member 2","SCY1 like pseudokinase 1","transmembrane protein 50B","amyloid beta precursor protein","DOP1 leucine zipper like protein B","URB1 ribosome biogenesis homolog","AKT serine/threonine kinase 1","interleukin 19","epithelial membrane protein 3","SUMO1 activating enzyme subunit 1","netrin 5","lemur tyrosine kinase 3","gem nuclear organelle associated protein 7","Cbl proto-oncogene C","WT1 interacting protein","ADAM metallopeptidase with thrombospondin type 1 motif 10","arginyl aminopeptidase like 1","calpain 10","myosin IF","endogenous retrovirus group K3 member 1","NLR family pyrin domain containing 12","calcium voltage-gated channel auxiliary subunit gamma 8","zinc finger protein 787","translocase of inner mitochondrial membrane 29","coactivator associated arginine methyltransferase 1","ecotropic viral integration site 5 like","solute carrier family 47 member 1","proteasome 20S subunit beta 6","sialic acid binding Ig like lectin 10","zinc finger protein 473","family with sequence similarity 71 member E1","ribosomal protein S11","ribosomal protein L13a","cytosolic thiouridylase subunit 1","nitric oxide synthase interacting protein","reticulocalbin 3","zinc finger protein 614","solute carrier family 2 member 5","arginine-glutamic acid dipeptide repeats","membrane metalloendopeptidase like 1","cilia and flagella associated protein 74","PR/SET domain 16","chymotrypsin like elastase 2A","peptidyl arginine deiminase 3","forkhead associated phosphopeptide binding domain 1","peptidyl arginine deiminase 1","EPH receptor A2","Rho guanine nucleotide exchange factor 19","EF-hand domain family member D2","peroxisomal biogenesis factor 14","phosphogluconate dehydrogenase","myomesin 3","SH3 domain binding glutamate rich protein like 3","connector enhancer of kinase suppressor of Ras 1","ribosomal protein L11","interleukin 22 receptor subunit alpha 1","zinc finger protein 593","chromosome 1 open reading frame 216","KIAA0319 like","eva-1 homolog B","DMRT like family A2","polo like kinase 4","mitogen-activated protein kinase kinase kinase 6","ficolin 3","GPN-loop GTPase 2","synaptotagmin like 1","WD and tetratricopeptide repeats 1","NBPF member 3","heparan sulfate proteoglycan 2","integrin subunit beta 3 binding protein","SERPINE1 mRNA binding protein 1","BCL10 immune signaling adaptor","cellular communication network factor 1","protein kinase cAMP-activated catalytic subunit beta","phosphatidylinositol glycan anchor biosynthesis class K","tubulointerstitial nephritis antigen like 1","antizyme inhibitor 2","ribosomal protein S8","kinesin family member 2C","protein tyrosine phosphatase receptor type F","bestrophin 4","MOB kinase activator 3C","cytochrome P450 family 4 subfamily B member 1","transmembrane protein 61","LIM domain only 4","metal response element binding transcription factor 2","solute carrier family 44 member 3","immunoglobulin superfamily member 3","zinc finger protein 697","CTTNBP2 N-terminal like","striatin interacting protein 1","proteasome 20S subunit alpha 5","chromosome 1 open reading frame 162","CD53 molecule","cadherin EGF LAG seven-pass G-type receptor 2","integrin subunit alpha 10","G protein-coupled receptor 161","aldehyde dehydrogenase 9 family member A1","ATPase Na+/K+ transporting subunit beta 1","TOR signaling pathway regulator","NME/NM23 family member 7","pogo transposable element derived with KRAB domain","mitochondrial pyruvate carrier 2","cellular repressor of E1A stimulated genes 1","DDB1 and CUL4 associated factor 6","T-box transcription factor 19","uridine-cytidine kinase 2","transmembrane and coiled-coil domains 1","POU class 2 homeobox 1","immunoglobulin like domain containing receptor 2","microsomal glutathione S-transferase 3","adenylate cyclase 10","COP1 E3 ubiquitin ligase","nectin cell adhesion molecule 4","ubiquitin-fold modifier conjugating enzyme 1","protoporphyrinogen oxidase","Fc fragment of IgG receptor IIa","NUF2 component of NDC80 kinetochore complex","regulator of G protein signaling 5","succinate dehydrogenase complex subunit C","prefoldin subunit 2","nuclear receptor subfamily 1 group I member 3","ubiquitin specific peptidase 21","proline rich mitotic checkpoint control factor","Fc receptor like 5","ribosomal RNA adenine dimethylase domain containing 1","mitochondrial ribosomal protein L24","phosphatidylinositol glycan anchor biosynthesis class M","interferon stimulated exonuclease gene 20 like 2","cellular retinoic acid binding protein 2","heparin binding growth factor","ABL proto-oncogene 2, non-receptor tyrosine kinase","xenotropic and polytropic retrovirus receptor 1","regulator of G protein signaling 16","torsin 1A interacting protein 1","hemicentin 1","ral guanine nucleotide dissociation stimulator like 1","lysophospholipase like 1","prune exopolyphosphatase 1","RAR related orphan receptor C","tuftelin 1","splicing factor 3b subunit 4","extracellular matrix protein 1","zinc finger protein 687","threonyl-tRNA synthetase 2, mitochondrial","cingulin","sorting nexin 27","SET domain bifurcated histone lysine methyltransferase 1","ADAMTS like 4","MCL1 apoptosis regulator, BCL2 family member","cathepsin K","regulatory factor X5","phosphatidylinositol 4-kinase beta","phosphatidylinositol-4-phosphate 5-kinase type 1 alpha","acidic nuclear phosphoprotein 32 family member E","MINDY lysine 48 deubiquitinase 1","annexin A9","selenium binding protein 1","ceramide synthase 2","endosulfine alpha","semaphorin 6C","mitochondrial ribosomal protein L9","aryl hydrocarbon receptor nuclear translocator","pogo transposable element derived with ZNF domain","chromosome 1 open reading frame 56","ornithine decarboxylase antizyme 3","HORMA domain containing 1","golgi phosphoprotein 3 like","GA binding protein transcription factor subunit beta 2","synaptotagmin 14","potassium voltage-gated channel subfamily H member 1","denticleless E3 ubiquitin protein ligase homolog","dual specificity tyrosine phosphorylation regulated kinase 3","eukaryotic translation initiation factor 2D","integrator complex subunit 7","vasohibin 2","TATA-box binding protein associated factor, RNA polymerase I subunit A","SET and MYND domain containing 2","sushi domain containing 4","dual specificity phosphatase 10","tumor protein p53 binding protein 2","ATPase phospholipid transporting 8B2","filaggrin family member 2","cornulin","ADAM metallopeptidase domain 15","jumping translocation breakpoint","RAB13, member RAS oncogene family","S100 calcium binding protein A8","tropomyosin 3","nucleoporin 210 like","SNAP associated protein","solute carrier family 27 member 3","S100 calcium binding protein A7","ubiquitin associated protein 2 like","solute carrier family 39 member 1","HCLS1 associated protein X-1","cAMP responsive element binding protein 3 like 4","ephrin A3","potassium calcium-activated channel subfamily N member 3","chromosome 1 open reading frame 43","GATA zinc finger domain containing 2B","interleukin enhancer binding factor 2","Ras like without CAAX 1","integrator complex subunit 3","hyperpolarization activated cyclic nucleotide gated potassium channel 3","filaggrin","chromosome 1 open reading frame 131","polypeptide N-acetylgalactosaminyltransferase 2","tetratricopeptide repeat domain 13","saccharopine dehydrogenase (putative)","lysosomal trafficking regulator","mitogen-activated protein kinase kinase kinase 21","centrosomal protein 170","acid phosphatase 1","synaptosome associated protein 47","signal recognition particle 9","nuclear VCP like","SDE2 telomere maintenance homolog","delta 4-desaturase, sphingolipid 1","F-box protein 28","ADP ribosylation factor 1","cornichon family AMPA receptor auxiliary protein 4","inositol-trisphosphate 3-kinase B","guanylate kinase 1","CDC42 binding protein kinase alpha","cornichon family AMPA receptor auxiliary protein 3","chromosome 1 open reading frame 35","membrane bound O-acyltransferase domain containing 2","poly(ADP-ribose) polymerase 1","presenilin 2","pyrroline-5-carboxylate reductase 2","lamin B receptor","Wnt family member 9A","epoxide hydrolase 1","renin","SRY-box transcription factor 13","ethanolamine kinase 2","PTPRF interacting protein alpha 4","pleckstrin homology domain containing A6","protein tyrosine phosphatase non-receptor type 7","synaptotagmin 2","ADP ribosylation factor like GTPase 8A","odd-skipped related transcription factor 1","growth differentiation factor 7","protein disulfide isomerase family A member 6","ras homolog family member B","ATPase H+ transporting V1 subunit C2","heterogeneous nuclear ribonucleoprotein L like","galactose mutarotase","calmodulin-lysine N-methyltransferase","ATP binding cassette subfamily G member 8","EMAP like 4","calmodulin 2","ChaC cation transport regulator homolog 2","ribosomal protein S27a","WD repeat containing planar cell polarity effector","VPS54 subunit of GARP complex","ASXL transcriptional regulator 2","ETAA1 activator of ATR kinase","small nuclear ribonucleoprotein polypeptide G","abhydrolase domain containing 1","Meis homeobox 1","cytosolic iron-sulfur assembly component 1","zinc finger protein 514","small nuclear ribonucleoprotein U5 subunit 200","mitochondrial ribosomal protein S5","ankyrin repeat domain 53","TP53RK binding protein","N-acetyltransferase 8 (putative)","exocyst complex component 6B","sideroflexin 5","testis expressed 261","DEAQ-box RNA dependent ATPase 1","dual specificity phosphatase 11","ST6 beta-galactoside alpha-2,6-sialyltransferase 2","nephrocystin 1","mal, T cell differentiation protein like","threonine synthase like 2","RAS like proto-oncogene B","transmembrane protein 177","5'-nucleotidase domain containing 4","RAB, member of RAS oncogene family like 2A","solute carrier family 20 member 1","fibulin 7","zinc finger CCCH-type containing 8","lipoyltransferase 1","cyclic nucleotide gated channel subunit alpha 3","fumarylacetoacetate hydrolase domain containing 2B","AF4/FMR2 family member 3","UBX domain protein 4","speckle type BTB/POZ protein like","RNA polymerase II subunit D","AMMECR1 like","polypeptide N-acetylgalactosaminyltransferase 13","plakophilin 4","sodium voltage-gated channel alpha subunit 1","solute carrier family 4 member 10","secernin 3","lunapark, ER junction formation factor","zinc finger protein 385B","cell division cycle associated 7","ubiquitin protein ligase E3 component n-recognin 3","phosphatase, orphan 2","GULP PTB domain containing engulfment adaptor 1","family with sequence similarity 171 member B","heat shock protein family D (Hsp60) member 1","coiled-coil domain containing 150","methyltransferase like 21A","unc-80 homolog, NALCN channel complex subunit","parathyroid hormone 2 receptor","neurobeachin like 1","KAT8 regulatory NSL complex subunit 1 like","sperm associated antigen 16","ATP binding cassette subfamily A member 12","sulfatase modifying factor 1","rhomboid domain containing 1","atypical chemokine receptor 3","transient receptor potential cation channel subfamily M member 8","hes family bHLH transcription factor 6","ankyrin repeat and MYND domain containing 1","COP9 signalosome subunit 7B","DIS3 like 3'-5' exoribonuclease 2","FA complementation group D2","TAM41 mitochondrial translocator assembly and maintenance homolog","vestigial like family member 4","RAB5A, member RAS oncogene family","reticulophagy regulator family member 2","CTD small phosphatase 1","CCR4-NOT transcription complex subunit 9","membrane associated ring-CH-type finger 4","serine/threonine kinase 11 interacting protein","GDP-mannose pyrophosphorylase A","glutamate receptor interacting protein 2","ELL associated factor 1","contactin 4","dynein cytoplasmic 1 light intermediate chain 1","RNA binding motif single stranded interacting protein 3","glutamate decarboxylase like 1","oxysterol binding protein like 10","protein O-linked mannose N-acetylglucosaminyltransferase 2 (beta 1,4-)","atypical chemokine receptor 2","cysteine and serine rich nuclear protein 1","solute carrier family 25 member 38","integrin subunit alpha 9","solute carrier family 22 member 14","golgin A4","CTD small phosphatase like","IQ motif and Sec7 domain ArfGEF 1","cullin associated and neddylation dissociated 2 (putative)","ribosomal protein L32","protein tyrosine phosphatase receptor type G","interleukin 17 receptor D","SHQ1, H/ACA ribonucleoprotein assembly factor","solute carrier family 25 member 26","ubiquitin like modifier activating enzyme 3","ADP ribosylation factor like GTPase 6 interacting protein 5","TATA element modulatory factor 1","leucine rich repeats and immunoglobulin like domains 1","leucine rich repeats and transmembrane domains 1","LIM domains containing 1","zinc finger protein 660","NFKB inhibitor zeta","collagen type VIII alpha 1 chain","neurexophilin and PC-esterase domain family member 3","myosin heavy chain 15","pleckstrin homology like domain family B member 2","abhydrolase domain containing 10","transgelin 3","phospholipase A1 member A","RAB, member of RAS oncogene family like 3","ADP-ribosylarginine hydrolase","immunoglobulin superfamily member 11","autophagy related 3","nuclear receptor subfamily 1 group I member 2","BOC cell adhesion associated, oncogene regulated","SRP receptor subunit beta","transmembrane protein 108","mediator complex subunit 12L","eukaryotic translation initiation factor 2A","aldehyde dehydrogenase 1 family member L1","oxysterol binding protein like 11","transient receptor potential cation channel subfamily C member 1","neutral cholesterol ester hydrolase 1","family with sequence similarity 86 member B2","LIM domain containing preferred translocation partner in lipoma","transmembrane protein 44","rubicon autophagy regulator","aminomethyltransferase","T cell leukemia translocation altered","nicolin 1","DDB1 and CUL4 associated factor 1","mesencephalic astrocyte derived neurotrophic factor","syntaxin binding protein 5 like","ELL associated factor 2","immunoglobulin like domain containing receptor 1","transmembrane 4 L six family member 19","mucin 4, cell surface associated","slit guidance ligand 2","eukaryotic translation initiation factor 2B subunit epsilon","diacylglycerol kinase theta","factor interacting with PAPOLA and CPSF1","solute carrier family 26 member 1","Ly1 antibody reactive","centromere protein C","corin, serine peptidase","ATPase phospholipid transporting 10D (putative)","OCIA domain containing 2","solute carrier family 10 member 6","stearoyl-CoA desaturase 5","placenta associated 8","enolase-phosphatase 1","tRNA methyltransferase 10A","kelch like family member 8","synuclein alpha","PIGY upstream reading frame","TBC1 domain containing kinase","calcium/calmodulin dependent protein kinase II delta","CDGSH iron sulfur domain 2","DNA damage inducible transcript 4 like","ankyrin 2","TRAF interacting protein with forkhead associated domain","spermatogenesis associated 5","fatty acid binding protein 2","cyclin A2","methyltransferase like 14","ubiquitin specific peptidase 53","SET domain containing 7, histone lysine methyltransferase","nuclear assembly factor 1 ribonucleoprotein","membrane associated ring-CH-type finger 1","ribosomal protein S3A","ring finger protein 175","platelet derived growth factor C","carbonyl reductase 4","glycine receptor alpha 3","cytochrome P450 family 4 subfamily V member 2","rhophilin associated tail protein 1 like","NADH:ubiquinone oxidoreductase subunit S6","membrane associated ring-CH-type finger 6","cadherin 18","ADAM metallopeptidase with thrombospondin type 1 motif 16","steroid 5 alpha-reductase 1","myosin X","OTU deubiquitinase with linear linkage specificity like","ribosomal protein L37","S-phase kinase associated protein 2","oncostatin M receptor","UDP glycosyltransferase family 3 member A1","polo like kinase 2","granzyme A","phosphoinositide-3-kinase regulatory subunit 1","LHFPL tetraspan subfamily member 2","single stranded DNA binding protein 2","betaine--homocysteine S-methyltransferase","ankyrin repeat domain 31","IQ motif containing GTPase activating protein 2","RAS p21 protein activator 1","limb and CNS expressed 1","gypsy retrotransposon integrase 1","diphosphoinositol pentakisphosphate kinase 2","peptidylglycine alpha-amidating monooxygenase","B double prime 1, subunit of RNA polymerase III transcription initiation factor IIIB","general transcription factor IIH subunit 2","solute carrier family 30 member 5","basic transcription factor 3","F-box and leucine rich repeat protein 17","TNF alpha induced protein 8","fem-1 homolog C","COMM domain containing 10","autophagy related 12","multiple EGF like domains 10","Yip1 domain family member 5","Rho GTPase activating protein 26","C-X-C motif chemokine ligand 14","leukocyte cell derived chemotaxin 2","solute carrier family 25 member 48","DEAD-box helicase 46","ring finger protein 145","gamma-aminobutyric acid type A receptor beta2 subunit","F-box protein 38","serine peptidase inhibitor, Kazal type 7 (putative)","prenylcysteine oxidase 1 like","glycine receptor alpha 1","TNFAIP3 interacting protein 1","G3BP stress granule assembly factor 1","zinc finger protein 300","NEDD4 binding protein 3","NHP2 ribonucleoprotein","required for meiotic nuclear division 5 homolog B","biorientation of chromosomes in cell division 1","complexin 2","teneurin transmembrane protein 2","potassium calcium-activated channel subfamily M regulatory beta subunit 1","family with sequence similarity 50 member B","myosin light chain kinase family member 4","family with sequence similarity 217 member A","TBC1 domain family member 7","phenylalanyl-tRNA synthetase 2, mitochondrial","glucose-fructose oxidoreductase domain containing 1","CDK5 regulatory subunit associated protein 1 like 1","leucine rich repeat transmembrane neuronal 2","zinc finger matrin-type 2","kelch like family member 3","doublecortin domain containing 2","tripartite motif containing 7","tripartite motif containing 41","HIG1 hypoxia inducible domain family member 2A","family with sequence similarity 193 member B","phospholipase A2 group VII","TNF receptor superfamily member 21","ring finger protein 44","methylmalonyl-CoA mutase","docking protein 3","activator of basal transcription 1","protein phosphatase 1 regulatory subunit 18","dishevelled associated activator of morphogenesis 2","DNA primase subunit 2","muscular LMNA interacting protein","lengsin, lens protein with glutamine synthetase domain","FYVE, RhoGEF and PH domain containing 2","signal peptide, CUB domain and EGF like domain containing 3","anoctamin 7","t-complex-associated-testis-expressed 1","ribosomal protein L7 like 1","NFKB inhibitor epsilon","cytochrome P450 family 39 subfamily A member 1","trophoblast glycoprotein","interleukin 1 receptor associated kinase 1 binding protein 1","pleckstrin homology domain interacting protein","MMS22 like, DNA repair protein","failed axon connections homolog, metaxin like GST domain containing","proline rich nuclear receptor coactivator 1","peptidase M20 domain containing 2","arginyl-tRNA synthetase 2, mitochondrial","Scm polycomb group protein like 4","TBC1 domain family member 32","ring finger protein 217","Rho GTPase activating protein 18","ABRA C-terminal like","solute carrier family 18 member B1","mitochondrial fission regulator 2","solute carrier family 2 member 12","SNF2 histone linker PHD RING helicase","androgen induced 1","dynein light chain Tctex-type 1","TIAM Rac1 associated GEF 2","transmembrane protein 181","PARN like, ribonuclease domain containing 1","WT1 associated protein","zinc finger MYM-type containing 4","acidic residue methyltransferase 1","von Willebrand factor D and EGF domains","G protein subunit alpha 12","chromosome 7 open reading frame 50","sidekick cell adhesion molecule 1","CCZ1 homolog B, vacuolar protein trafficking and biogenesis associated","chromosome 7 open reading frame 26","RB associated KRAB zinc finger","cAMP responsive element binding protein 5","epidermal growth factor receptor","cell division cycle associated 5","insulin like growth factor binding protein 3","purine rich element binding protein B","malate dehydrogenase 2","POM121 and ZP3 fusion","nipsnap homolog 2","chaperonin containing TCP1 subunit 6A","phosphoserine phosphatase","zinc finger protein 92","ataxin 7 like 1","transmembrane protein 168","ankyrin repeat and SOCS box containing 15","microtubule associated protein 11","solute carrier family 12 member 9","GRB10 interacting GYF protein 1","tripartite motif containing 4","methylphosphate capping enzyme","transmembrane protein 209","ATP/GTP binding protein like 3","zinc finger CCCH-type containing, antiviral 1 like","transmembrane protein 140","tousled like kinase 2","EPH receptor A1","nucleolar protein with MIF4G domain 1","canopy FGF signaling regulator 1","non-SMC condensin II complex subunit G2","neuroligin 4 X-linked","shroom family member 2","RAB19, member RAS oncogene family","LUC7 like 2, pre-mRNA splicing factor","DENN domain containing 2A","collectrin, amino acid transport regulator","SH3 domain containing kinase binding protein 1","transmembrane protein 47","LanC like 3","synaptotagmin like 5","calcium/calmodulin dependent serine protein kinase","lysine demethylase 6A","spindlin family member 2A","moesin","cyclin B3","histone deacetylase 8","solute carrier family 16 member 2","divergent protein kinase domain 2B","zinc finger protein 157","zinc finger protein 182","carbohydrate sulfotransferase 7","KRAB box domain containing 4","NADH:ubiquinone oxidoreductase subunit B11","zinc finger protein 41","zinc finger MYM-type containing 3","TATA-box binding protein associated factor 1","G protein-coupled receptor 174","non-POU domain containing octamer binding","coiled-coil domain containing 120","lysophosphatidic acid receptor 4","EBP cholestenol delta-isomerase","O-linked N-acetylglucosamine (GlcNAc) transferase","sorting nexin 12","integrin subunit beta 1 binding protein 2","interleukin 2 receptor subunit gamma","germ cell nuclear acidic peptidase","zinc finger protein 711","diaphanous related formin 2","nuclear RNA export factor 3","phosphoribosyl pyrophosphate synthetase 1","RPA1 related single stranded DNA binding protein, X-linked","FERM and PDZ domain containing 3","dedicator of cytokinesis 11","immunoglobulin superfamily member 1","glypican 3","RNA binding motif protein X-linked","microcephalin 1","malignant fibrous histiocytoma amplified sequence 1","F-box protein 25","NAD(P) dependent steroid dehydrogenase-like","zinc finger protein 185 with LIM domain","centrin 2","ribosomal protein L10","chondroitin sulfate N-acetylgalactosaminyltransferase 1","ATPase H+ transporting V1 subunit B2","coiled-coil domain containing 25","homeobox containing 1","gonadotropin releasing hormone 1","bridging integrator 3","docking protein 2","solute carrier family 25 member 37","charged multivesicular body protein 7","dedicator of cytokinesis 5","steroidogenic acute regulatory protein","pyridoxal phosphate binding protein","ER lipid raft associated 2","syntrophin gamma 1","ST18 C2H2C-type zinc finger transcription factor","regulator of G protein signaling 20","transforming acidic coiled-coil containing protein 1","golgin A7","phospholipid phosphatase 5","GINS complex subunit 4","nuclear receptor binding SET domain protein 3","DnaJ heat shock protein family (Hsp40) member C5 beta","alcohol dehydrogenase iron containing 1","mitochondrial ribosomal protein S28","peripheral myelin protein 2","lactamase beta 2","PR/SET domain 14","telomeric repeat binding factor 1","ribosomal protein L7","solute carrier family 26 member 7","ATPase H+ transporting V0 subunit d2","syntabulin","dihydropyrimidinase","metadherin","LDL receptor related protein 12","estrogen receptor binding site associated antigen 9","R-spondin 2","RNA polymerase II subunit K","mal, T cell differentiation protein 2 (gene/pseudogene)","eukaryotic translation initiation factor 3 subunit H","UTP23 small subunit processome component","NADH:ubiquinone oxidoreductase subunit B9","TatD DNase domain containing 1","family with sequence similarity 83 member A","gasdermin C","family with sequence similarity 135 member B","zinc finger protein 7","Rho GTPase activating protein 39","solute carrier family 39 member 4","nicotinate phosphoribosyltransferase","very low density lipoprotein receptor","adenylate kinase 3","ubiquitin like with PHD and ring finger domains 2","nuclear factor I B","perilipin 2","HAUS augmin like complex subunit 6","cyclin dependent kinase inhibitor 2B","cyclin dependent kinase inhibitor 2A","C9orf72-SMCR8 complex subunit","interferon kappa","zinc finger CCHC-type containing 7","F-box protein 10","sigma non-opioid intracellular receptor 1","COBW domain containing 5","centrosomal protein 78","neurotrophic receptor tyrosine kinase 2","IDNK gluconokinase","SHC adaptor protein 3","AU RNA binding methylglutaconyl-CoA hydratase","major facilitator superfamily domain containing 14B","aminopeptidase O (putative)","zinc finger protein 462","INTS3 and NABP interacting protein","UDP-glucose ceramide glucosyltransferase","sorting nexin family member 30","stomatin","gelsolin","mitochondrial ribosome recycling factor","nuclear receptor subfamily 6 group A member 1","crumbs cell polarity complex component 2","aminolevulinate dehydratase","astrotactin 2","WD repeat domain 31","DNA polymerase epsilon 3, accessory subunit","surfeit 4","globoside alpha-1,3-N-acetylgalactosaminyltransferase 1 (FORS blood group)","SURF1 cytochrome c oxidase assembly factor","surfeit 2","surfeit 6","mediator complex subunit 22","REX4 homolog, 3'-5' exonuclease","ribosomal protein L7a","general transcription factor IIIC subunit 5","ankyrin repeat and SOCS box containing 6","prostaglandin E synthase 2","N-terminal Xaa-Pro-Lys N-methyltransferase 1","CDKN1A interacting zinc finger protein 1","solute carrier family 25 member 25","SH3 domain containing GRB2 like, endophilin B2","mitoguardin 2","prostaglandin E synthase","lipocalin 2","leucine rich repeat and sterile alpha motif containing 1","hemicentin 2","G protein-coupled receptor 107","PAXX non-homologous end joining factor","inositol polyphosphate-5-phosphatase E","SEC16 homolog A, endoplasmic reticulum export factor","diphthamide biosynthesis 7","notch receptor 1","calcium voltage-gated channel subunit alpha1 B","NACC family member 2","proline and serine rich 2","USP6 N-terminal like","COMM domain containing 3","methionine sulfoxide reductase B2","decaprenyl diphosphate synthase subunit 1","family with sequence similarity 171 member A1","MINDY lysine 48 deubiquitinase 3","transmembrane protein 236","Ras suppressor protein 1","ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 6","par-3 family cell polarity regulator","zinc finger E-box binding homeobox 1","family with sequence similarity 13 member C","nuclear receptor binding factor 2","APOBEC1 complementation factor","retinal G protein coupled receptor","RNA polymerase III subunit A","HECT and RLD domain containing E3 ubiquitin protein ligase 4","leucine rich melanocyte differentiation associated","calcium/calmodulin dependent protein kinase II gamma","adipogenesis regulatory factor","glutamate dehydrogenase 1","5-hydroxytryptamine receptor 7","ribonuclease P/MRP subunit p30","FRA10A associated CGG repeat 1","adducin 3","DnaJ heat shock protein family (Hsp40) member B12","eukaryotic translation initiation factor 4E binding protein 2","neuropeptide FF receptor 1","pleckstrin homology domain containing S1","transcription factor 7 like 2","marker of proliferation Ki-67","cytochrome P450 family 17 subfamily A member 1","fucose mutarotase","leucine rich repeat containing 27","mitochondrial ribosome associated GTPase 1","polyamine oxidase","glutathione S-transferase omega 1","TATA-box binding protein associated factor 5","PPARG related coactivator 1","inositol 1,4,5-trisphosphate receptor interacting protein","cyclin and CBS domain divalent metal cation transport mediator 2","programmed cell death 11","ADAM metallopeptidase domain 12","regulator of G protein signaling 10","BTB domain containing 10","adrenomedullin","growth arrest specific 2","solute carrier family 5 member 12","lin-7 homolog C, crumbs cell polarity complex component","inner mitochondrial membrane peptidase subunit 1","serum amyloid A4, constitutive","post-GPI attachment to proteins 2","terminal uridylyl transferase 1, U6 snRNA-specific","secretoglobin family 1A member 1","synaptotagmin 8","zinc finger protein 214","zinc finger protein 215","hydroxysteroid 17-beta dehydrogenase 12","APAF1 interacting protein","peptidase domain containing associated with muscle regeneration 1","diacylglycerol kinase zeta","eukaryotic translation initiation factor 3 subunit M","tankyrase 1 binding protein 1","glycine-N-acyltransferase","serpin family G member 1","structure specific recognition protein 1","solute carrier family 43 member 1","protein tyrosine phosphatase receptor type J","chromosome 11 open reading frame 49","ADP ribosylation factor GTPase activating protein 2","CUGBP Elav-like family member 1","heat shock protein nuclear import factor hikeshi","coiled-coil domain containing 81","sestrin 3","endonuclease domain containing 1","coiled-coil domain containing 82","kelch like family member 35","teneurin transmembrane protein 4","serpin family H member 1","calpain 5","integrator complex subunit 4","p21 (RAC1) activated kinase 1","ribosomal protein S3","zinc finger CCCH-type containing 12C","tetratricopeptide repeat domain 12","neural cell adhesion molecule 1","dopamine receptor D2","chromosome 11 open reading frame 52","5-hydroxytryptamine receptor 3B","nuclear protein, coactivator of histone transcription","ATM serine/threonine kinase","aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl transferase","galactosidase beta 1 like 2","SLX4 interacting protein","late endosomal/lysosomal adaptor, MAPK and MTOR activator 1","prolyl 4-hydroxylase subunit alpha 3","glutamate ionotropic receptor kainate type subunit 4","suppression of tumorigenicity 14","hypoxia up-regulated 1","ADAM metallopeptidase domain 33","lysine acetyltransferase 14","triokinase and FMN cyclase","metastasis associated 1 family member 2","transmembrane protein 138","fatty acid desaturase 1","transmembrane channel like 2","retinal outer segment membrane protein 1","EMAP like 3","inner centromere protein","phospholipase C eta 2","cleavage and polyadenylation specific factor 7","membrane spanning 4-domains A2","beta-1,3-glucuronyltransferase 3","EI24 autophagy associated transmembrane protein","coiled-coil domain containing 15","checkpoint kinase 1","fasciculation and elongation protein zeta 1","endothelial cell adhesion molecule","kirre like nephrin family adhesion molecule 3","myelin protein zero like 2","sodium voltage-gated channel beta subunit 2","SID1 transmembrane family member 2","transmembrane protein 25","transgelin","COMM domain containing 7","chromosome 20 open reading frame 144","DSN1 component of MIS12 kinetochore complex","suppressor of glucose, autophagy associated 1","cyclic nucleotide binding domain containing 2","cadherin 22","LSM family member 14B","YTH N6-methyladenosine RNA binding protein 1","Cdk5 and Abl enzyme substrate 2","LTO1 maturation factor of ABCE1","solute carrier family 22 member 9","tRNA phosphotransferase 1","nudix hydrolase 22","fermitin family member 3","phospholipase C beta 3","mitochondrial ribosomal protein L49","CDC42 effector protein 2","FAU ubiquitin like and ribosomal protein S30 fusion","transmembrane 7 superfamily member 2","VPS51 subunit of GARP complex","T-box transcription factor 6","protein phosphatase 4 catalytic subunit","aldolase, fructose-bisphosphate A","TLC domain containing 3B","double C2 domain alpha","HIRA interacting protein 3","TAO kinase 2","transmembrane protein 219","high mobility group AT-hook 2","matrix metallopeptidase 3","connector enhancer of kinase suppressor of Ras 2","contactin 5","killer cell lectin like receptor F1","C-type lectin domain family 1 member A","mohawk homeobox","membrane palmitoylated protein 7","integrin subunit beta 1","protocadherin related 15","cardiotrophin 1","CWC15 spliceosome associated protein homolog","Fc fragment of IgG receptor Ia","AT-rich interaction domain 5B","cadherin 8","defective in cullin neddylation 1 domain containing 2","transmembrane and coiled-coil domains 3","transmembrane protein 218","TIR domain containing adaptor protein","EEF1A lysine methyltransferase 1","large tumor suppressor kinase 2","Sin3A associated protein 18","adhesion G protein-coupled receptor L3","KIAA1328","family with sequence similarity 124 member A","MIA SH3 domain ER export factor 2","histamine N-methyltransferase","LY6/PLAUR domain containing 1","LY6/PLAUR domain containing 6B","programmed cell death 4","adrenoceptor alpha 2A","glycoprotein M6A","WD repeat domain 17","spermatogenesis associated 4","vascular endothelial growth factor C","coiled-coil domain containing 102B","CD226 molecule","carnosine dipeptidase 1","fibrous sheath interacting protein 1","discs large MAGUK scaffold protein 2","coiled-coil domain containing 83","regulator of G protein signaling 18","serine protease 23","myotubularin related protein 12","protein phosphatase 1 regulatory inhibitor subunit 1C","chromosome 11 open reading frame 53","chaperonin containing TCP1 subunit 5","ATP synthase c subunit lysine N-methyltransferase","dedicator of cytokinesis 1","DIX domain containing 1","dihydrolipoamide S-acetyltransferase","PIH1 domain containing 2","NKAP domain containing 1","translocase of inner mitochondrial membrane 8 homolog B","interleukin 18","6-pyruvoyltetrahydropterin synthase","phosphatidylinositol-5-phosphate 4-kinase type 2 alpha","chromosome 2 open reading frame 50","FRAS1 related extracellular matrix 2","forkhead box O1","cysteine rich transmembrane BMP regulator 1","SEC24 homolog D, COPII coat complex component","ATP binding cassette subfamily B member 9","Rab interacting lysosomal protein like 2","DEAH-box helicase 37","ubiquitin C","inositol 1,4,5-trisphosphate receptor type 1","serine protease 53","solute carrier family 7 member 11","nocturnin","enkurin, TRPC channel interacting protein","calcium voltage-gated channel auxiliary subunit alpha2delta 4","decapping mRNA 1B","calcium voltage-gated channel subunit alpha1 C","thyroid hormone receptor beta","N-glycanase 1","3-oxoacyl-ACP synthase, mitochondrial","UEV and lactate/malate dehyrogenase domains","transmembrane protein 86A","chromosome 12 open reading frame 45","transmembrane protein 263","BTB domain containing 11","ubiquitin protein ligase E3B","ankyrin 3","inositol polyphosphate multikinase","RAD9 checkpoint clamp component B","phospholipase B domain containing 2","discs large MAGUK scaffold protein 5","methionine adenosyltransferase 1A","solute carrier family 2 member 13","glucoside xylosyltransferase 1","twinfilin actin binding protein 1","disco interacting protein 2 homolog C","eukaryotic translation initiation factor 4E","membrane associated guanylate kinase, WW and PDZ domain containing 1","testis expressed 30","casein kinase 1 gamma 3","serum response factor binding protein 1","A-kinase anchoring protein 6","neuronal PAS domain protein 3","family with sequence similarity 177 member A1","MAP3K12 binding inhibitory protein 1","mirror-image polydactyly 1","exostosin glycosyltransferase 2","transmembrane protein 18","potassium channel tetramerization domain containing 14","NADH:ubiquinone oxidoreductase subunit C2","malic enzyme 3","ADAM metallopeptidase with thrombospondin type 1 motif 12","nucleotide binding protein like","NIMA related kinase 7","FER tyrosine kinase","VPS33B interacting protein, apical-basolateral polarity regulator, spe-39 homolog","ankyrin repeat domain 50","UPF2 regulator of nonsense mediated mRNA decay","cell division cycle 123","sodium channel and clathrin linker 1","chromosome 4 open reading frame 33","FERM domain containing 4A","protein tyrosine phosphatase receptor type O","epidermal growth factor receptor pathway substrate 8","acyl-CoA dehydrogenase family member 8","thymocyte nuclear protein 1","VPS26, retromer complex component B","non-SMC condensin II complex subunit D3","vesicle transport through interaction with t-SNAREs 1A","quinoid dihydropteridine reductase","family with sequence similarity 160 member B1","anoctamin 4","testis expressed 9","queuine tRNA-ribosyltransferase accessory subunit 2","metabolism of cobalamin associated A","zinc finger protein 827","nuclear receptor subfamily 3 group C member 2","aldo-keto reductase family 1 member C2","VENT homeobox","ADAM metallopeptidase domain 8","inter-alpha-trypsin inhibitor heavy chain 2","Kin17 DNA and RNA binding protein","phosphatidylinositol glycan anchor biosynthesis class F","ankyrin and armadillo repeat containing","inositol polyphosphate-1-phosphatase","major facilitator superfamily domain containing 6","ring finger protein 144A","ArfGAP with SH3 domain, ankyrin repeat and PH domain 2","ADAM metallopeptidase domain 17","Fli-1 proto-oncogene, ETS transcription factor","potassium inwardly rectifying channel subfamily J member 1","transmembrane protein 45B","WW and C2 domain containing 2","centromere protein U","acyl-CoA synthetase long chain family member 1","solute carrier family 25 member 4","antagonist of mitotic exit network 1 homolog","BICD cargo adaptor 1","salvador family WW domain containing protein 1","coiled-coil domain containing 122","stress associated endoplasmic reticulum protein family member 2","NBAS subunit of NRZ tethering complex","zinc finger protein 385D","GUF1 homolog, GTPase","solute carrier family 35 member F4","gamma-aminobutyric acid type A receptor alpha2 subunit","sacsin molecular chaperone","coiled-coil domain containing 175","centromere protein J","F-box protein 4","transmembrane protein 267","C-C motif chemokine ligand 28","poly(ADP-ribose) polymerase family member 8","CDK2 associated cullin domain 1","dystonin","BEN domain containing 6","TIA1 cytotoxic granule associated RNA binding protein like 1","BCL2 associated athanogene 3","glycosyltransferase 1 domain containing 1","schwannomin interacting protein 1","melanin concentrating hormone receptor 2","potassium voltage-gated channel subfamily E regulatory subunit 4","adaptor related protein complex 1 subunit sigma 3","RAB GTPase activating protein 1 like","coiled-coil domain containing 74B","TLC domain containing 4","mitotic spindle organizing protein 2B","tubulin alpha 3e","family with sequence similarity 168 member B","protein tyrosine phosphatase non-receptor type 14","alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase","transmembrane protein 163","G-patch domain containing 11","heat shock protein family B (small) member 8","gem nuclear organelle associated protein 6","transmembrane protein 178A","ORC ubiquitin ligase 1","cysteinyl leukotriene receptor 2","ADP ribosylation factor like GTPase 11","Ras like without CAAX 2","SET binding protein 1","ADP ribosylation factor like GTPase 14 effector protein","ectopic P-granules autophagy protein 5 homolog","proline-serine-threonine phosphatase interacting protein 2","ATP synthase F1 subunit alpha","HAUS augmin like complex subunit 1","chromosome 18 open reading frame 25","SPC25 component of NDC80 kinetochore complex","pyruvate dehydrogenase kinase 1","phosphodiesterase 3B","transcription factor 7 like 1","trans-golgi network protein 2","SH2 domain containing 6","U2AF homology motif kinase 1","autophagy related 10","POC5 centriolar protein","SPARC (osteonectin), cwcv and kazal like domains proteoglycan 1","family with sequence similarity 151 member B","transcriptional adaptor 1","CWF19 like cell cycle control factor 2","junction mediating and regulatory protein, p53 cofactor","homer scaffold protein 1","X-ray repair cross complementing 4","boule homolog, RNA binding protein","zinc finger protein 547","zinc finger protein 773","zinc finger protein 776","zinc finger protein 256","suppressor of variegation 3-9 homolog 2","DNA cross-link repair 1C","oleoyl-ACP hydrolase","ribonuclease P/MRP subunit p38","N-myristoyltransferase 2","zinc finger and SCAN domain containing 1","zinc finger protein 837","ubiquitin specific peptidase 12","coiled-coil domain containing 50","calcium/calmodulin dependent protein kinase IV","tripartite motif containing 36","ZFP36 ring finger protein like 2","poly(A) specific ribonuclease subunit PAN3","pleckstrin homology, MyTH4 and FERM domain containing H2","phosphofructokinase, muscle","transmembrane protein 123","glutamate ionotropic receptor AMPA type subunit 4","immunoglobulin superfamily member 10","sperm flagellar 2","SPARC like 1","matrix extracellular phosphoglycoprotein","muscleblind like splicing regulator 1","calcyphosine like","NAD kinase 2, mitochondrial","glycerol-3-phosphate dehydrogenase 1 like","gap junction protein alpha 1","cyclin O","solute carrier family 30 member 6","pelota mRNA surveillance and ribosome rescue factor","RAS guanyl releasing protein 3","secretion associated Ras related GTPase 1B","cation channel sperm associated 3","G protein-coupled receptor 180","Tctex1 domain containing 1","WD repeat domain 78","ankyrin repeat domain 22","FERM, ARH/RhoGEF and pleckstrin domain protein 1","interferon induced protein with tetratricopeptide repeats 5","solute carrier family 16 member 12","pantothenate kinase 1","PR/SET domain 8","bone morphogenetic protein 3","heterogeneous nuclear ribonucleoprotein D like","hematopoietically expressed homeobox","utrophin","glutamate metabotropic receptor 1","protein tyrosine phosphatase receptor type K","geranylgeranyl diphosphate synthase 1","zinc finger protein 117","lamin tail domain containing 1","MARVEL domain containing 2","RAD17 checkpoint clamp loader component","mediator complex subunit 21","procollagen-lysine,2-oxoglutarate 5-dioxygenase 2","neurensin 1","janus kinase and microtubule interacting protein 1","Zic family member 1","adhesion G protein-coupled receptor A3","carboxypeptidase B1","SREK1 interacting protein 1","CWC27 spliceosome associated cyclophilin","major histocompatibility complex, class I-related","signal recognition particle 19","centromere protein H","chromodomain Y like","calcium regulated heat stable protein 1","tektin 5","B cell scaffold protein with ankyrin repeats 1","thioredoxin domain containing 11","DAB adaptor protein 2","acyl-CoA oxidase like","BCL2 like 11","anaphase promoting complex subunit 1","calpastatin","short coiled-coil protein","calmegin","centrin 3","SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 5","synaptonemal complex protein 2 like","bone morphogenetic protein 6","Ras association domain family member 3","heterogeneous nuclear ribonucleoprotein U","RAN binding protein 2","AT-hook containing transcription factor 1","MER proto-oncogene, tyrosine kinase","transmembrane protein 87B","nuclear receptor subfamily 4 group A member 2","coiled-coil domain containing 148","phospholipase A2 receptor 1","RNA binding motif single stranded interacting protein 1","sodium voltage-gated channel alpha subunit 3","CD96 molecule","solute carrier family 25 member 27","adhesion G protein-coupled receptor F1","family with sequence similarity 49 member B","ArfGAP with SH3 domain, ankyrin repeat and PH domain 1","trafficking protein particle complex 8","family with sequence similarity 81 member B","INO80 complex subunit C","lysophosphatidylcholine acyltransferase 1","pleckstrin homology and RhoGEF domain containing G4B","NmrA like redox sensor 1","UBA like domain containing 1","chromosome 16 open reading frame 89","transmembrane protein 251","inhibitor of growth family member 1","sperm acrosome associated 7","ADP-ribosylhydrolase like 1","CKLF like MARVEL transmembrane domain containing 7","F-box and leucine rich repeat protein 2","upstream binding protein 1","required for meiotic nuclear division 5 homolog A","CD8a molecule","ribose 5-phosphate isomerase A","leucine rich adaptor protein 1 like","CNKSR family member 3","general transcription factor IIE subunit 1","craniofacial development protein 1","zinc finger DHHC-type containing 7","family with sequence similarity 92 member B","chromosome 7 open reading frame 31","transmembrane serine protease 11D","JAZF zinc finger 1","c-Maf inducing protein","SPHK1 interactor, AKAP domain containing","potassium inwardly rectifying channel subfamily J member 16","phosphotyrosine interaction domain containing 1","thyroid hormone receptor interactor 12","F-box protein 36","CCAAT enhancer binding protein gamma","potassium channel tetramerization domain containing 15","zinc finger protein 599","mucolipin 2","leucine rich repeat LGI family member 4","dimethylarginine dimethylaminohydrolase 1","splicing regulatory glutamic acid and lysine rich protein 1","chromodomain helicase DNA binding protein 1","ankyrin repeat and fibronectin type III domain containing 1","diacylglycerol kinase epsilon","heparan sulfate 2-O-sulfotransferase 1","musashi RNA binding protein 2","calcium voltage-gated channel auxiliary subunit alpha2delta 1","zinc finger containing ubiquitin peptidase 1","glycerophosphodiester phosphodiesterase domain containing 1","NUS1 dehydrodolichyl diphosphate synthase subunit","semaphorin 3D","protein phosphatase 2 regulatory subunit B'epsilon","GRB2 related adaptor protein","adenylate kinase 5","calcium binding tyrosine phosphorylation regulated","impact RWD domain protein","ankyrin repeat domain 29","succinate dehydrogenase complex assembly factor 4","carbohydrate sulfotransferase 9","Thy-1 cell surface antigen","dynein axonemal assembly factor 1","chromosome 16 open reading frame 74","tubulin folding cofactor E like","junctophilin 3","ANKH inorganic pyrophosphate transport regulator","OTU deubiquitinase with linear linkage specificity","ubiquitin associated and SH3 domain containing B","roundabout guidance receptor 3","transforming growth factor beta regulator 1","reticulophagy regulator 1","cadherin 12","translocase of outer mitochondrial membrane 70","ABI family member 3 binding protein","angiopoietin 1","phosphatidylinositol transfer protein cytoplasmic 1","coiled-coil and C2 domain containing 1B","ceramide synthase 3","protein kinase C alpha","leucine rich repeat kinase 1","centrosomal protein 112","galactose-3-O-sulfotransferase 2","ATP binding cassette subfamily A member 9","ATP binding cassette subfamily A member 6","ATP binding cassette subfamily A member 10","ATP binding cassette subfamily A member 5","ectonucleotide pyrophosphatase/phosphodiesterase 3","chromosome 4 open reading frame 19","ubiquitin C-terminal hydrolase L1","MIA SH3 domain ER export factor 3","dispatched RND transporter family member 1","TRAF2 and NCK interacting kinase","family with sequence similarity 167 member A","nei like DNA glycosylase 2","phosphoglucomutase 5","Wnt family member 3A","obscurin, cytoskeletal calmodulin and titin-interacting RhoGEF","LON peptidase N-terminal domain and ring finger 1","tripartite motif containing 11","ENAH actin regulator","centriole, cilia and spindle associated protein","ankyrin repeat, SAM and basic leucine zipper domain containing 1","SH3 domain containing ring finger 1","guanylate binding protein 5","BUB3 mitotic checkpoint protein","coiled-coil domain containing 173","matrix metallopeptidase 21","divergent protein kinase domain 1A","ATP synthase membrane subunit c locus 3","contactin associated protein like 3B","serine and arginine rich splicing factor 12","PDZ and LIM domain 3","sorbin and SH3 domain containing 2","elongin C","lymphocyte antigen 96","CXADR Ig-like cell adhesion molecule","BTG anti-proliferation factor 3","chromosome 21 open reading frame 91","chondrolectin","neural cell adhesion molecule 2","L3MBTL histone methyl-lysine binding protein 4","phosphodiesterase 1C","RAB guanine nucleotide exchange factor 1","mitochondrial ribosomal protein L39","junctional adhesion molecule 2","ATP synthase peripheral stalk subunit F6","GA binding protein transcription factor subunit alpha","ADAM metallopeptidase with thrombospondin type 1 motif 1","tRNA splicing endonuclease subunit 2","schlafen family member 13","XPC complex subunit, DNA damage recognition and repair factor","coiled-coil domain containing 174","FYVE, RhoGEF and PH domain containing 5","folliculin","diphthamide biosynthesis 3","oxidoreductase NAD binding domain containing 1","phospholipase C like 2","CXXC finger protein 1","spindle and kinetochore associated complex subunit 1","protein phosphatase 4 regulatory subunit 1","APC down-regulated 1","piezo type mechanosensitive ion channel component 2","metallophosphoesterase 1","ubiquitin specific peptidase 43","RAB6B, member RAS oncogene family","essential meiotic structure-specific endonuclease 1","EPH receptor B1","acyl-CoA synthetase short chain family member 1","ankyrin repeat domain 40","zinc finger protein 18","VOPP1 WW domain binding protein","septin 14","apolipoprotein O like","cytochrome P450 family 2 subfamily U member 1","radial spoke head 10 homolog B","F-box and leucine rich repeat protein 18","contactin associated protein like 5","prominin 2","adenylate kinase 9","Kruppel like factor 10","protein tyrosine phosphatase receptor type N2","antizyme inhibitor 1","ATPase H+ transporting V1 subunit C1","phosphatidylinositol-4,5-bisphosphate 4-phosphatase 2","OTU deubiquitinase 6B","cyclin dependent kinase 19","general transcription factor IIIC subunit 6","tetratricopeptide repeat domain 39B","1-acylglycerol-3-phosphate O-acyltransferase 5","MMS19 homolog, cytosolic iron-sulfur assembly component","phosphatidylinositol 4-kinase type 2 alpha","MARVEL domain containing 1","zinc finger FYVE-type containing 27","golgin A7 family member B","G protein-coupled receptor 78","tRNA methyltransferase 44 homolog","solute carrier family 25 member 28","heat shock protein family A (Hsp70) member 13","SAM domain, SH3 domain and nuclear localization signals 1","ubiquitin specific peptidase 25","GRAM domain containing 2B","zinc finger CCHC-type containing 10","chromosome 16 open reading frame 87","Mov10 RISC complex RNA helicase","ras homolog family member C","protein phosphatase, Mg2+/Mn2+ dependent 1J","diazepam binding inhibitor, acyl-CoA binding protein","solute carrier family 16 member 1","HEAT repeat containing 3","nucleolar protein interacting with the FHA domain of MKI67","OXA1L mitochondrial inner membrane protein","solute carrier family 7 member 7","La ribonucleoprotein 1, translational regulator","CCR4-NOT transcription complex subunit 8","glutamate ionotropic receptor AMPA type subunit 1","leucine rich repeats and guanylate kinase domain containing","SET domain containing 9","MIER family member 3","nucleoporin 205","zinc finger with KRAB and SCAN domains 2","chromosome 9 open reading frame 85","phosphoinositide-3-kinase adaptor protein 1","RNA binding motif protein 45","titin","V-set and immunoglobulin domain containing 4","protein disulfide isomerase family A member 4","lysine demethylase 8","PDZ domain containing 9","otoancorin","potassium channel tetramerization domain containing 18","family with sequence similarity 126 member B","flagellum associated containing coiled-coil domains 1","C2 calcium dependent domain containing 6","transmembrane protein 237","frizzled class receptor 7","sperm associated antigen 17","DEP domain containing MTOR interacting protein","formin 2","ring finger protein 20","PPARG coactivator 1 beta","engulfment and cell motility 1","solute carrier family 26 member 2","LSM11, U7 small nuclear RNA associated","mediator complex subunit 7","stabilizer of axonemal microtubules 1","Ras related GTP binding A","solute carrier family 24 member 2","2-phosphoxylose phosphatase 1","RAS p21 protein activator 2","required for meiotic nuclear division 1 homolog","Src like adaptor","transmembrane BAX inhibitor motif containing 4","VHL binding protein 1","RAB39B, member RAS oncogene family","chloride intracellular channel 2","AF4/FMR2 family member 2","mitochondrial calcium uptake family member 3","glutamate receptor interacting protein 1","VPS37A subunit of ESCRT-I","pleckstrin and Sec7 domain containing 3","carnosine N-methyltransferase 1","mitochondrial calcium uniporter","ELM2 and Myb/SANT domain containing 1","cilia and flagella associated protein 70","G protein subunit alpha 14","FAM161 centrosomal protein B","G protein subunit alpha q","G protein-coupled receptor 61","adenosine kinase","potassium calcium-activated channel subfamily M alpha 1","basic leucine zipper ATF-like transcription factor","deoxycytidine kinase","ADAM metallopeptidase with thrombospondin type 1 motif 3","ALX homeobox 3","dpy-19 like 4","NADH:ubiquinone oxidoreductase complex assembly factor 6","DNA damage regulated autophagy modulator 2","chromosome 8 open reading frame 37","protein phosphatase with EF-hand domain 2","cilia and flagella associated protein 161","ADAMTS like 3","ADP-ribosyltransferase 3","WASP homolog associated with actin, golgi membranes and microtubules","C-X-C motif chemokine ligand 13","N-6 adenine-specific DNA methyltransferase 1","RWD domain containing 2B","ubiquitin specific peptidase 16","chaperonin containing TCP1 subunit 8","MAP3K7 C-terminal like","N(alpha)-acetyltransferase 11, NatA catalytic subunit","BTB domain and CNC homolog 1","claudin 8","TIAM Rac1 associated GEF 1","SR-related CTD associated factor 4","retinitis pigmentosa GTPase regulator","cyclin dependent kinase 20","polycomb group ring finger 6","ankyrin repeat domain 9","SWI5 dependent homologous recombination repair protein 1","sideroflexin 2","ATP synthase membrane subunit 6.8PL","fucosyltransferase 6","tudor domain containing 9","fibroblast growth factor 18","protocadherin 1","SH3 domain containing ring finger 2","ubiquinol-cytochrome c reductase binding protein","mitochondrial transcription termination factor 3","phosphatidylserine synthase 1","protein phosphatase 2 regulatory subunit Bbeta","ribosomal protein L30","potassium voltage-gated channel modifier subfamily S member 2","family with sequence similarity 122C","Suv3 like RNA helicase","family with sequence similarity 122B","eukaryotic translation elongation factor 1 alpha 1","F-box protein 43","hexokinase 1","trypsin domain containing 1","PHD finger protein 6","CD109 molecule","nodal growth differentiation factor","ubiquitin conjugating enzyme E2 L6","zinc finger DHHC-type containing 5","mediator complex subunit 19","zinc finger AN1-type containing 3","neuroplastin","lysine acetyltransferase 6B","sterile alpha motif domain containing 8","RAB11 family interacting protein 1","UTP14A small subunit processome component","apoptosis inducing factor mitochondria associated 1","mitogen-activated protein kinase 13","BCL2 associated athanogene 4","membrane spanning 4-domains A1","TBC1 domain family member 31","WDYHV motif containing 1","ATPase family AAA domain containing 2","F-box protein 32","NSE2 (MMS21) homolog, SMC5-SMC6 complex SUMO ligase","zinc finger protein 689","proline rich 14","fibrosin","ferric chelate reductase 1","phosphorylase kinase catalytic subunit gamma 2","major facilitator superfamily domain containing 14A","SAS-6 centriolar assembly protein","integrin subunit alpha D","Zic family member 3","mitochondrial assembly of ribosomal large subunit 1","VPS8 subunit of CORVET complex","galactokinase 2","LHFPL tetraspan subfamily member 4","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 7","MPV17 mitochondrial inner membrane protein like","BUB1 mitotic checkpoint serine/threonine kinase B","phosphodiesterase 6D","eukaryotic translation initiation factor 4A2","bromodomain and PHD finger containing 1","RNA pseudouridine synthase D3","TatD DNase domain containing 2","ghrelin and obestatin prepropeptide","SEC13 homolog, nuclear pore and COPII coat complex component","exo/endonuclease G","N-terminal asparagine amidase","nicotinamide nucleotide adenylyltransferase 2","zinc finger FYVE-type containing 9","ATPase plasma membrane Ca2+ transporting 2","solute carrier family 6 member 1","SMG1 nonsense mediated mRNA decay associated PI3K related kinase","FCH and mu domain containing endocytic adaptor 2","RNA binding protein, mRNA processing factor","transmembrane protein 171","TIMP metallopeptidase inhibitor 4","synapsin II","neuregulin 1","odr-4 GPCR localization factor homolog","carnitine palmitoyltransferase 2","NECAP endocytosis associated 2","LDL receptor related protein 8","PAX interacting protein 1","STEAP2 metalloreductase","single stranded DNA binding protein 3","claudin 12","matrix metallopeptidase 14","frizzled class receptor 1","GATA zinc finger domain containing 1","sushi domain containing 3","transmembrane p24 trafficking protein 6","C-type lectin domain family 18 member A","dehydrogenase/reductase 4","chromosome 1 open reading frame 158","C-type lectin domain family 18 member C","DEAD-box helicase 19B","ST3 beta-galactoside alpha-2,3-sialyltransferase 2","fucose kinase","interleukin 34","dehydrogenase/reductase 1","calcium voltage-gated channel subunit alpha1 D","arylsulfatase L","KIT proto-oncogene, receptor tyrosine kinase","HYDIN axonemal central pair apparatus protein","aminoadipate-semialdehyde dehydrogenase","zinc finger protein 19","calcium voltage-gated channel auxiliary subunit alpha2delta 3","ring finger protein 111","cyclin B2","family with sequence similarity 81 member A","myosin IE","adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1","actin filament associated protein 1 like 1","TSC22 domain family member 3","VPS26 endosomal protein sorting factor C","dual specificity tyrosine phosphorylation regulated kinase 1A","potassium inwardly rectifying channel subfamily J member 6","potassium inwardly rectifying channel subfamily J member 15","ETS proto-oncogene 2, transcription factor","tetraspanin 18","lebercilin LCA5 like","solute carrier family 35 member B2","transmembrane protein 164","MX dynamin like GTPase 1","cAMP responsive element binding protein 3 like 1","C2 calcium dependent domain containing 2","TGF-beta activated kinase 1 (MAP3K7) binding protein 3","solute carrier family 38 member 10","chromosome 9 open reading frame 43","PALM2 and AKAP2 fusion","zinc finger protein 618","diacylglycerol kinase iota","transmembrane protein 268","SVOP like","sorting nexin 22","ubinuclein 2","B-Raf proto-oncogene, serine/threonine kinase","solute carrier family 34 member 2","aggrecan","proteasome assembly chaperone 3","calcium binding protein 1","WD repeat domain 19","solute carrier family 37 member 3","adaptor related protein complex 3 subunit sigma 2","formin like 2","GRB2 associated regulator of MAPK1 subtype 2","signal peptide peptidase like 3","dihydropyrimidinase like 5","dynein regulatory complex subunit 1","RAB28, member RAS oncogene family","peroxiredoxin like 2B","TNF receptor superfamily member 14","pantothenate kinase 4 (inactive)","multiple EGF like domains 11","chromosome 12 open reading frame 43","peroxisomal biogenesis factor 10","retention in endoplasmic reticulum sorting receptor 1","Rap associating with DIL domain","SKI proto-oncogene","WD repeat domain, phosphoinositide interacting 2","low density lipoprotein receptor adaptor protein 1","ArfGAP with GTPase domain, ankyrin repeat and PH domain 1","keratinocyte associated protein 3","ankyrin repeat domain 61","platelet activating factor acetylhydrolase 2","solute carrier family 30 member 2","BRISC and BRCA1 A complex member 2","WD repeat domain 66","mitochondrial ribosomal protein L17","dual specificity phosphatase 2","grainyhead like transcription factor 3","UBX domain protein 11","NLR family pyrin domain containing 14","protein tyrosine phosphatase domain containing 1","polypeptide N-acetylgalactosaminyltransferase 14","NCK adaptor protein 1","4-hydroxyphenylpyruvate dioxygenase","rhophilin Rho GTPase binding protein 1","tumor protein p63 regulated 1 like","leucine rich repeat containing 43","peroxiredoxin like 2C","xanthine dehydrogenase","XK related 8","cyclin and CBS domain divalent metal cation transport mediator 4","EYA transcriptional coactivator and phosphatase 3","DAZ interacting zinc finger protein 1 like","FA complementation group C","muscle RAS oncogene homolog","WASP family member 2","abhydrolase domain containing 3","extended synaptotagmin 3","Fas apoptotic inhibitory molecule","terminal nucleotidyltransferase 5B","calsyntenin 2","collectin subfamily member 12","ring finger protein 207","cullin 4B","G protein-coupled receptor 153","solute carrier family 13 member 3","G protein-coupled receptor associated sorting protein 2","rhomboid like 2","activator of transcription and developmental regulator AUTS2","shroom family member 4","H2B clustered histone 5","H4 clustered histone 8","microtubule interacting and trafficking domain containing 1","eukaryotic translation initiation factor 5B","RIB43A domain with coiled-coils 1","thymosin beta 15B","ciliogenesis associated TTC17 interacting protein","CCR4-NOT transcription complex subunit 11","potassium voltage-gated channel subfamily B member 1","tetraspanin 33","neuregulin 2","adenosylhomocysteinase like 2","beta-1,4-galactosyltransferase 5","CD1d molecule","spermatogenesis associated 2","CD1c molecule","family with sequence similarity 86 member C1","dynein axonemal heavy chain 3","CD1e molecule","neutrophil cytosolic factor 1","TSR2 ribosome maturation factor","protein phosphatase 1 regulatory subunit 9A","zinc finger CCCH-type containing 18","zinc finger AN1-type containing 2B","glycerophosphodiester phosphodiesterase domain containing 5","dynein cytoplasmic 1 intermediate chain 1","transmembrane p24 trafficking protein 4","protein phosphatase 1 regulatory subunit 15B","coatomer protein complex subunit gamma 2","EMSY transcriptional repressor, BRCA2 interacting","glycerol-3-phosphate acyltransferase 4","polycystin 1 like 1, transient receptor potential channel interacting","zinc finger and SCAN domain containing 12","transgelin 2","ETS transcription factor ELK4","SLAM family member 8","solute carrier family 45 member 3","dual specificity phosphatase 23","ring finger protein 166","NBL1, DAN family BMP antagonist","5-hydroxytryptamine receptor 6","F11 receptor","upstream transcription factor 1","spermatogenesis associated 2 like","nitrilase 1","death effector domain containing","zinc finger protein 276","nucleophosmin/nucleoplasmin 2","ectodysplasin A","von Willebrand factor A domain containing 5B1","cytidine deaminase","PTEN induced kinase 1","beta-1,4-galactosyltransferase 3","dematin actin binding protein","ADAM metallopeptidase with thrombospondin type 1 motif 4","family with sequence similarity 160 member B2","NADH:ubiquinone oxidoreductase core subunit S2","Fc fragment of IgE receptor Ig","apolipoprotein A2","translocase of outer mitochondrial membrane 40 like","cell cycle and apoptosis regulator 2","Wnt family member 9B","cache domain containing 1","CDC42 small effector 2","Rap guanine nucleotide exchange factor 6","erythrocyte membrane protein band 4.1","MIS18 kinetochore protein A","ALG8 alpha-1,3-glucosyltransferase","F-box and WD repeat domain containing 5","cilia and flagella associated protein 298","synaptojanin 1","PAX3 and PAX7 binding protein 1","interferon alpha and beta receptor subunit 2","mitochondrial ribosomal protein L10","interferon gamma receptor 2","phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, phosphoribosylaminoimidazole synthetase","SON DNA binding protein","downstream neighbor of SON","synaptic vesicle glycoprotein 2A","ladinin 1","stanniocalcin 1","troponin I1, slow skeletal type","cysteine and glycine rich protein 1","complement C1q C chain","ATP synthase membrane subunit c locus 1","regulator of calcineurin 1","ubiquitin conjugating enzyme E2 Z","circadian associated repressor of transcription","SNF8 subunit of ESCRT-II","chloride intracellular channel 6","coiled-coil domain containing 24","RUNX family transcription factor 1","insulin like growth factor 2 mRNA binding protein 1","carbonyl reductase 1","carbonyl reductase 3",null,"gap junction protein delta 2","MORC family CW-type zinc finger 3","chromatin assembly factor 1 subunit B","SIM bHLH transcription factor 2","holocarboxylase synthetase","golgin A6 family member A","signal peptide, CUB domain and EGF like domain containing 1","Rho GTPase activating protein 27","ADP dependent glucokinase","parathymosin","phospholipase A2 group IVD","peptidyl arginine deiminase 4","adiponectin receptor 1","cytochrome b5 reductase 1","proteasome 26S subunit, non-ATPase 4","ATPase cation transporting 13A2","meiosis 1 associated protein","proteasome 20S subunit beta 4","BTG anti-proliferation factor 2","hexokinase 2","complement C1r","CUGBP Elav-like family member 3","aldehyde dehydrogenase 4 family member A1","StAR related lipid transfer domain containing 9","thioesterase superfamily member 4","trichohyalin","ubiquitin protein ligase E3 component n-recognin 1","autocrine motility factor receptor","mediator complex subunit 8","ral guanine nucleotide dissociation stimulator like 4","ring finger and SPRY domain containing 1","coiled-coil domain containing 17","GC-rich promoter binding protein 1 like 1","NEDD8 activating enzyme E1 subunit 1","transmembrane protein 69","adhesion G protein-coupled receptor G5","dynein regulatory complex subunit 7","angiotensin I converting enzyme","testis, prostate and placenta expressed","EF-hand calcium binding domain 14","spondin 2","coiled-coil-helix-coiled-coil-helix domain containing 6","C-terminal binding protein 1","leucine rich repeat containing 36","tubulin polymerization promoting protein family member 3","zinc finger DHHC-type containing 1","ATPase H+ transporting V0 subunit d1","zinc finger FYVE-type containing 28","capping protein regulator and myosin 1 linker 2","chromosome 16 open reading frame 86","prolactin induced protein","family with sequence similarity 131 member B","regulator of G protein signaling 12","protein serine kinase H1","zyxin","ABR activator of RhoGEF and GTPase","LY6/PLAUR domain containing 5","coiled-coil domain containing 117","zinc finger protein 230","coiled-coil domain containing 107","zinc finger protein 222","natriuretic peptide receptor 2","zinc finger protein 221","zinc finger protein 233","zinc finger protein 235","glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase","TNF receptor superfamily member 13C","olfactory receptor family 3 subfamily A member 3","Rho GTPase activating protein 35","prostaglandin I2 receptor","calmodulin 3","DNA fragmentation factor subunit alpha","coiled-coil domain containing 28B","IQ motif containing C","transmembrane protein 234","BSD domain containing 1","zinc finger and BTB domain containing 8A","ATPase family AAA domain containing 3B","SSU72 homolog, RNA polymerase II CTD phosphatase","ubiquitin conjugating enzyme E2 J2","zinc finger protein 362","fibronectin type III domain containing 5","C3 and PZP like alpha-2-macroglobulin domain containing 8","nuclear receptor subfamily 2 group F member 6","ankyrin repeat and LEM domain containing 1","coiled-coil domain containing 58","vacuolar ATPase assembly factor VMA21","kalirin RhoGEF kinase","ATP binding cassette subfamily G member 1","trefoil factor 3","trefoil factor 1","transmembrane serine protease 3","ubiquitin associated and SH3 domain containing A","radial spoke head component 1","solute carrier family 37 member 1","phosphodiesterase 9A","WD repeat domain 4","NADH:ubiquinone oxidoreductase subunit V3","PBX/knotted 1 homeobox 1","cystathionine beta-synthase","U2 small nuclear RNA auxiliary factor 1","heat shock transcription factor 2 binding protein","ribosomal RNA processing 1B","pyridoxal kinase","glucose-6-phosphate dehydrogenase","cystatin B","ribosomal RNA processing 1","1-acylglycerol-3-phosphate O-acyltransferase 3","trafficking protein particle complex 10","GRB2 associated binding protein 3","glutamine amidotransferase like class 1 domain containing 3A","inducible T cell costimulator ligand","autoimmune regulator","cilia and flagella associated protein 410","zinc finger protein 66","leucine rich repeat containing 3","integrin subunit beta 2","family with sequence similarity 207 member A","ral guanine nucleotide dissociation stimulator","spermatogenesis and centriole associated 1 like","lanosterol synthase","vav guanine nucleotide exchange factor 2","minichromosome maintenance complex component 3 associated protein","chromosome 21 open reading frame 58","pericentrin","disco interacting protein 2 homolog A","S100 calcium binding protein B","protein arginine methyltransferase 2","zinc finger protein 208","ADAM metallopeptidase with thrombospondin type 1 motif 13","calcium channel flower domain containing 1","solute carrier family 2 member 6","zinc finger protein 761","chromosome 9 open reading frame 116","zinc finger protein 714","G protein signaling modulator 1","chromosome 19 open reading frame 47","cilia and flagella associated protein 157","torsin family 2 member A","ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 6","SH3KBP1 binding protein 1","retinol dehydrogenase 13","zyg-11 related cell cycle regulator","zinc finger DHHC-type containing 12","protein kinase N3","spectrin beta, non-erythrocytic 4","cytochrome c oxidase subunit 6B2","transmembrane protein 190","phospholipid phosphatase 7 (inactive)","TAO kinase 1","mediator complex subunit 27","death effector domain containing 2","SIK family kinase 3","myelin protein zero like 3","junction adhesion molecule like","NIMA related kinase 8","TLC domain containing 1","proprotein convertase subtilisin/kexin type 7","scaffold attachment factor B","CD3g molecule","S100 calcium binding protein A1","chromatin target of PRMT1","C-X-C motif chemokine receptor 5","zinc finger and BTB domain containing 7B","flavin adenine dinucleotide synthetase 1","SHC adaptor protein 1","VPS11 core subunit of CORVET and HOPS complexes","NLR family member X1","adenosine deaminase RNA specific","interleukin 6 receptor","ubiquitin conjugating enzyme E2 Q1","cholinergic receptor nicotinic beta 2 subunit","CREB regulated transcription coactivator 2","anoctamin 10","farnesyl diphosphate synthase","RUN and SH3 domain containing 1","family with sequence similarity 189 member B","progestin and adipoQ receptor family member 6","polyamine modulated factor 1","solute carrier family 25 member 44","lamin A/C","C-C motif chemokine receptor 5 (gene/pseudogene)","neurobeachin like 2","coiled-coil domain containing 12","ubiquilin 4","myosin light chain 3","protein phosphatase 1 regulatory subunit 35","G-patch domain containing 4","leucine rich repeat containing 71","Fc receptor like 3","alpha-2-glycoprotein 1, zinc-binding","fibroblast growth factor receptor 4","nucleus accumbens associated 1","cytochrome P450 family 11 subfamily B member 1","hexokinase 3","lymphocyte antigen 6 family member K","immediate early response 2","zinc finger protein 394","cleavage and polyadenylation specific factor 4","lymphocyte antigen 6 family member E","VPS28 subunit of ESCRT-I","tonsoku like, DNA repair protein","PWWP domain containing 3A, DNA repair factor","RecQ like helicase 4","leucine rich repeat containing 14","zinc finger protein 333","protein phosphatase 1 regulatory subunit 16A","ORAI calcium release-activated calcium modulator 2","alkB homolog 4, lysine demethylase","SH2B adaptor protein 2","MRN complex interacting protein","sequestosome 1","alpha-1,3-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase B","ribosomal protein L8","mastermind like transcriptional coactivator 1","leucine rich repeats and WD repeat domain containing 1","F-box and leucine rich repeat protein 13","N-acyl phosphatidylethanolamine phospholipase D","secretoglobin family 3A member 1","proteasome 26S subunit, ATPase 2","CUGBP Elav-like family member 5","major facilitator superfamily domain containing 12","ubiquitin specific peptidase 41","YdjC chitooligosaccharide deacetylase homolog","dishevelled segment polarity protein 3","adaptor related protein complex 2 subunit mu 1","ATP binding cassette subfamily F member 3","phosphate cytidylyltransferase 1, choline, alpha","F-box protein 27","dermokine","U2 small nuclear RNA auxiliary factor 1 like 4","3-hydroxybutyrate dehydrogenase 1","NPHS1 adhesion molecule, nephrin","THAP domain containing 8","cytochrome c oxidase subunit 7A1","zinc finger protein 382","leucine rich repeat containing 56","plexin domain containing 1","post-GPI attachment to proteins phospholipase 3","IKAROS family zinc finger 3","glutamate ionotropic receptor NMDA type subunit 2C","ferredoxin reductase","SAP30 binding protein","acyl-CoA oxidase 1","phosphoribosyl pyrophosphate synthetase associated protein 1","cytoglobin","serine and arginine rich splicing factor 2","zinc finger protein 577","transmembrane protein 143","aldehyde dehydrogenase 16 family member A1","dermcidin","integrin subunit alpha 5","sialic acid binding Ig like lectin 11","zinc finger protein 385A","membrane palmitoylated protein 3","IZUMO family member 2","N-acetylglutamate synthase","LSM12 homolog","ankyrin repeat and SOCS box containing 16","ER membrane protein complex subunit 10","Josephin domain containing 2","SH3 and multiple ankyrin repeat domains 1","family with sequence similarity 171 member A2","DBF4 zinc finger B","phospholipase C delta 3","formin like 3","aquaporin 5","Rac GTPase activating protein 1","La ribonucleoprotein 4","general receptor for phosphoinositides 1 associated scaffold protein","ribonucleoprotein, PTB binding 1","SPC24 component of NDC80 kinetochore complex","inositol hexakisphosphate kinase 3","LEM domain containing 2","arachidonate 15-lipoxygenase","zinc finger protein 653","mediator complex subunit 11","C-X-C motif chemokine ligand 16","SLP adaptor and CSK interacting membrane protein","TNF superfamily member 13","SUMO specific peptidase 3","fibroblast growth factor 11","eukaryotic translation initiation factor 4A1","ribosomal protein L26","RNA polymerase III subunit K","small nuclear ribonucleoprotein U11/U12 subunit 25","WD repeat domain 90","jumonji domain containing 8","coiled-coil domain containing 78",null,"splA/ryanodine receptor domain and SOCS box containing 3","heparan sulfate-glucosamine 3-sulfotransferase 6","tubulin epsilon and delta complex 2","cyclin F","TBC1 domain family member 24","amidohydrolase domain containing 2","BICD family like cargo adaptor 2","progestin and adipoQ receptor family member 4","FLYWCH family member 2","zymogen granule protein 16B","zinc finger protein 75a","adenylate cyclase 9","SH3 and multiple ankyrin repeat domains 2","ClpB homolog, mitochondrial AAA ATPase chaperonin","neuraminidase 3","cytochrome b561 family member A3","protein phosphatase 1 regulatory subunit 32","asparaginase and isoaspartyl peptidase 1","UBX domain protein 1","LBH domain containing 1","tetratricopeptide repeat domain 9C","TATA-box binding protein associated factor 6 like","nuclear RNA export factor 1","syntaxin 5","solute carrier family 25 member 45","ribosomal protein L29","inter-alpha-trypsin inhibitor heavy chain 3","synoviolin 1","zinc finger protein like 1","ribosomal protein S6 kinase A4","LDL receptor related protein 5","two pore segment channel 2","cytochrome P450 family 4 subfamily A member 22","PDZK1 interacting protein 1","TAL bHLH transcription factor 1, erythroid differentiation factor","cytidine/uridine monophosphate kinase 1","BEN domain containing 5","cytochrome c oxidase assembly factor 7 (putative)","zyg-11 family member B, cell cycle regulator","CXXC motif containing zinc binding protein","mago homolog, exon junction complex subunit","acyl-CoA thioesterase 11","family with sequence similarity 151 member A","prolyl-tRNA synthetase 2, mitochondrial","lymphocyte expansion molecule","barttin CLCNK type accessory beta subunit","ubiquitin specific peptidase 24","phospholipid phosphatase 3","nucleolar protein 9","protein kinase AMP-activated catalytic subunit alpha 2","kelch like family member 21","zinc finger SWIM-type containing 5","glucocorticoid modulatory element binding protein 1","solute carrier family 45 member 1","selenoprotein N","adenylate kinase 4","Janus kinase 1","ribonucleoprotein, PTB binding 2","chymotrypsin C","leucine zipper and CTNNBIP1 domain containing","retinol binding protein 7","kinocilin","filamin binding LIM protein 1","solute carrier family 25 member 34","aldo-keto reductase family 7 member A3","dorsal inhibitory axon guidance protein","podoplanin","leucine rich repeat containing 38","dehydrogenase/reductase 3","matrilin 1","lysosomal protein transmembrane 5","syndecan 3","penta-EF-hand domain containing 1","syncoilin, intermediate filament protein","RB binding protein 4, chromatin remodeling factor","KIAA1522","testis specific serine kinase 3","transmembrane and coiled-coil domains 4","UBX domain protein 10","calcium/calmodulin dependent protein kinase II inhibitor 1","alkaline phosphatase, biomineralization associated","Wnt family member 4","tubulin tyrosine ligase like 10","sodium channel epithelial 1 delta subunit","matrix remodeling associated 8","FA core complex associated protein 20","multiple EGF like domains 6","coiled-coil domain containing 27","interleukin 23 receptor","chromosome 1 open reading frame 87","nuclear factor I A","OMA1 zinc metallopeptidase","Myb like, SWIRM and MPN domains 1","TM2 domain containing 1","ubiquitin specific peptidase 1","far upstream element binding protein 1","nexilin F-actin binding protein","DnaJ heat shock protein family (Hsp40) member B4","adhesion G protein-coupled receptor L4","leucine rich repeats and IQ motif containing 3","tRNA-yW synthesizing protein 3 homolog","sorting nexin 7","beta-1,3-galactosyltransferase 2","family with sequence similarity 102 member B","HEN methyltransferase 1","AKNA domain containing 1","chromosome 1 open reading frame 52","WD repeat domain 63","guanylate binding protein 2","ataxin 7 like 2","guanylate binding protein 4","zinc finger protein 326","growth factor independent 1 transcriptional repressor","amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase","exostosin like glycosyltransferase 2","solute carrier family 30 member 7","zinc finger protein 281","actin related protein 2/3 complex subunit 5","NLR family pyrin domain containing 3","zinc finger protein 496","tripartite motif containing 58","immunoglobulin superfamily member 8","discoidin domain receptor tyrosine kinase 2","proliferation and apoptosis adaptor protein 15","peroxisomal biogenesis factor 19","nicastrin","VANGL planar cell polarity protein 2","SLAM family member 6","olfactomedin like 2B","Fc fragment of IgG receptor IIIb","solute carrier family 9 member C2 (putative)","kelch domain containing 9","chromosome 1 open reading frame 74","feline leukemia virus subgroup C cellular receptor 1","family with sequence similarity 71 member A","activating transcription factor 3","RNA binding motif protein 15","DENN domain containing 2D","axonemal dynein light chain domain containing 1","immediate early response 5","sushi, nidogen and EGF like domains 1","3'(2'), 5'-bisphosphate nucleotidase 1","spermatogenesis associated 17","chromosome 1 open reading frame 115","BRO1 domain and CAAX motif containing","NBPF member 20","acid phosphatase 6, lysophosphatidic","kinesin family member 26B","transcription factor B2, mitochondrial","consortin, connexin sorting protein","protein phosphatase 1 regulatory subunit 21","kelch domain containing 8A","oxoeicosanoid receptor 1","3-hydroxyanthranilate 3,4-dioxygenase","beta-1,3-N-acetylgalactosaminyltransferase 2","MAPK activated protein kinase 2","Fc fragment of IgM receptor","polymeric immunoglobulin receptor","calpain 2","mitochondrial ribosomal protein L55","WD repeat domain 26","REL proto-oncogene, NF-kB subunit","pseudouridine synthase 10","peroxisomal biogenesis factor 13","KIAA1841","tripartite motif containing 17","raftlin family member 2","DISC1 scaffold protein","calpain 13","mediator of cell motility 1","dpy-30 histone methyltransferase complex regulatory subunit","tRNA-yW synthesizing protein 5","matrix AAA peptidase interacting protein 1","solute carrier family 66 member 3","ADP ribosylation factor like GTPase 5A","LRAT domain containing 1","potassium inwardly rectifying channel subfamily J member 3","clathrin heavy chain linker domain containing 1","dual specificity phosphatase 19","cilia and flagella associated protein 36","nucleoporin 35","coiled-coil domain containing 138","F-box protein 41","actin gamma 2, smooth muscle","WD repeat and coiled coil containing","structural maintenance of chromosomes 6","visinin like 1","coiled-coil domain containing 74A","H3.3 histone A","ankyrin repeat domain 30B like","coenzyme Q8A","solute carrier family 16 member 14","tektin 4","sarcoglycan beta","spermatogenesis associated 18","nitric oxide synthase trafficking","cilia and flagella associated protein 221","sphingosine-1-phosphate phosphatase 2","inhibin subunit beta B","Bardet-Biedl syndrome 5","SWI/SNF-related, matrix-associated actin-dependent regulator of chromatin, subfamily a, containing DEAD/H box 1","hematopoietic prostaglandin D synthase","PDZ and LIM domain 5","sperm tail PG-rich repeat containing 2","neuralized E3 ubiquitin protein ligase 3","regulation of nuclear pre-mRNA domain containing 2","ankyrin repeat domain 23","cathepsin S","msh homeobox 1","parkin coregulated like","BCL2 interacting protein like","C1q and TNF related 7","TNF alpha induced protein 8 like 2","LysM domain containing 1","sodium channel modifier 1","tropomodulin 4","vacuolar protein sorting 72 homolog","ERCC excision repair 3, TFIIH core complex helicase subunit","ring finger protein 149","interacts with SUPT6H, CTD assembly factor 1","bolA family member 3","CDC42 effector protein 3","S100 calcium binding protein A11","small proline rich protein 3","DExH-box helicase 57","small proline rich protein 2D","Rho GTPase activating protein 25","S100 calcium binding protein A9","S100 calcium binding protein A12","transforming growth factor alpha","cyclin Y like 1","frizzled class receptor 5","DDB1 and CUL4 associated factor 16","chromosome 1 open reading frame 189","glucosamine-6-phosphate deaminase 2","alkaline phosphatase, placental","progestin and adipoQ receptor family member 3","NIPA like domain containing 1","ANTXR cell adhesion molecule 2","helicase, POLQ like","mitochondrial ribosomal protein S18C","CGG triplet repeat binding protein 1","abraxas 1, BRCA1 A complex subunit","G protein-coupled receptor 155","death associated protein like 1","phosphomevalonate kinase","PBX homeobox interacting protein 1","claudin 1","pygopus family PHD finger 2","homeodomain interacting protein kinase 1","DC-STAMP domain containing 2","DC-STAMP domain containing 1","collagen type VI alpha 3 chain","innate immunity activator","YY1 associated protein 1","kelch repeat and BTB domain containing 8","EGF domain specific O-linked N-acetylglucosamine transferase","leiomodin 3","NAD(P)HX epimerase","protein O-glucosyltransferase 1","solute carrier family 22 member 15","ATPase Na+/K+ transporting subunit alpha 1","solute carrier family 15 member 2","eukaryotic translation initiation factor 4E family member 3","prokineticin 2","testis expressed 55","leucine rich repeat containing 58","follistatin like 1","leiomodin 1","E74 like ETS transcription factor 3","transmembrane protein 183A","transmembrane protein 169","insulin like growth factor binding protein 7","tripartite motif containing 46","keratinocyte associated protein 2","C-X-C motif chemokine receptor 1","actin related protein 2/3 complex subunit 2","chaperonin containing TCP1 subunit 3","transmembrane protein 79","signal sequence receptor subunit 2","ring finger protein 25","serine/threonine kinase 36","NIMA related kinase 10","coiled-coil domain containing 141","cell proliferation regulating inhibitor of protein phosphatase 2A","eomesodermin","CWC22 spliceosome associated protein homolog","5-azacytidine induced 2","transforming growth factor beta receptor 2","ankyrin repeat and zinc finger peptidyl tRNA hydrolase 1","histone deacetylase 11","galactosidase beta 1 like","STT3 oligosaccharyltransferase complex catalytic subunit B","coiled-coil-helix-coiled-coil-helix domain containing 4","neurofascin","Fc receptor like 1","shugoshin 2","serpin family I member 1","cytoplasmic linker associated protein 2","succinate-CoA ligase alpha subunit","NUAK family kinase 2","protein kinase C iota","myeloid cell nuclear differentiation antigen","pyrin and HIN domain family member 1","interferon gamma inducible protein 16","absent in melanoma 2","EF-hand domain family member B","eukaryotic translation initiation factor 5A2","ribosomal protein L22 like 1","protein phosphatase, Mg2+/Mn2+ dependent 1L","islet cell autoantigen 1 like","cytotoxic T-lymphocyte associated protein 4","inducible T cell costimulator","RING1 and YY1 binding protein","protein phosphatase 4 regulatory subunit 2","CD200 receptor 1","GTP binding protein 8 (putative)","nucleolus and neural progenitor protein","spindle and centriole associated protein 1","coiled-coil domain containing 191","calcium dependent secretion activator","CDP-diacylglycerol synthase 1","WD repeat and FYVE domain containing 3","cytochrome c oxidase assembly factor COX18","protein tyrosine phosphatase non-receptor type 13","synaptoporin","chromosome 3 open reading frame 49","chromosome 4 open reading frame 36","THO complex 7","ataxin 7","proteasome 26S subunit, non-ATPase 6","prickle planar cell polarity protein 2","ADAM metallopeptidase with thrombospondin type 1 motif 9","protein phosphatase, Mg2+/Mn2+ dependent 1K","glutamate rich 6","guanine monophosphate synthase","TCDD inducible poly(ADP-ribose) polymerase","cyclin L1","pentraxin 3","HESX homeobox 1","sarcolemma associated protein","ribosomal protein L9","small integral membrane protein 14","ribonuclease P/MRP subunit p14","abhydrolase domain containing 6","deoxyribonuclease 1 like 3","chromosome 3 open reading frame 67","RNA binding motif protein 47","amyloid beta precursor protein binding family B member 2","interleukin 17 receptor E","interleukin 17 receptor C","cysteine rich with EGF like domains 1","proline rich transmembrane protein 3","procollagen C-endopeptidase enhancer 2","U2 snRNP associated SURP domain containing","myotubularin related protein 14","tetratricopeptide repeat domain 14","C-X-C motif chemokine ligand 3","C-X-C motif chemokine ligand 5","methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2 like","C-X-C motif chemokine ligand 1","ring finger and CHY zinc finger domain containing 1","phospholipid scramblase 2","coiled-coil domain containing 158","carboxypeptidase A3","glycogenin 1","HPS3 biogenesis of lysosomal organelles complex 2 subunit 1","DNA topoisomerase II binding protein 1","receptor like tyrosine kinase","SNF related kinase","transcription factor 23","zinc finger protein 513","solute carrier family 4 member 1 adaptor protein","phospholipase B1","speedy/RINGO cell cycle regulator family member A","KIAA1143","kinesin family member 15","transglutaminase 4","WD repeat domain 43","zinc finger DHHC-type containing 3","CUB domain containing protein 1","solute carrier family 6 member 20","leucine zipper transcription factor like 1","FYVE and coiled-coil domain containing 1","C-C motif chemokine receptor 1","leucine rich repeat containing 2","elongator acetyltransferase complex subunit 6","F-box protein 40","deltex E3 ubiquitin ligase 3L","zinc finger protein 148","nicotinamide nucleotide adenylyltransferase 3","small integral membrane protein 12","zinc finger MYM-type containing 6","transmembrane protein adipocyte associated 1","YEATS domain containing 2","zinc finger CCCH-type containing 12A","MYST/Esa1 associated factor 6","Smad nuclear interacting protein 1","dynein axonemal light intermediate chain 1","RNA polymerase II subunit H","Kruppel like factor 15","cilia and flagella associated protein 100","lipase H","transmembrane protein 41A","ribophorin I","SUMO specific peptidase 2","intraflagellar transport 122","replication factor C subunit 4","ribosomal protein L39 like","BRCA1 associated protein 1","transketolase","protein kinase C delta","RFT1 homolog","Scm like with four mbt domains 1","G protein nucleolar 3","polybromo 1","UV stimulated scaffold protein A","transcription activation suppressor","Rho guanine nucleotide exchange factor 3","stem-loop binding protein","LDL receptor related protein associated protein 1","zinc finger DHHC-type containing 19","solute carrier family 51 alpha subunit","UBX domain protein 7","ring finger protein 168","phosphatidylinositol glycan anchor biosynthesis class X","melanotransferrin","S100 calcium binding protein P","actin binding LIM protein family member 2","exonuclease 5","claudin 19","chromosome 1 open reading frame 50","erythroblast membrane associated protein (Scianna blood group)","zinc finger protein 691","aminoacyl tRNA synthetase complex interacting multifunctional protein 1","sphingomyelin synthase 2","methionyl aminopeptidase 1","DnaJ heat shock protein family (Hsp40) member B14","H2A.Z variant histone 1","endomucin","solute carrier family 9 member B1","solute carrier family 9 member B2","3-hydroxybutyrate dehydrogenase 2","progesterone receptor membrane component 2","cell division cycle 25A","zinc finger protein 589","F-box and WD repeat domain containing 12","plexin B1","coiled-coil domain containing 51","ATR interacting protein","shisa family member 5","sprouty RTK signaling antagonist 1","bassoon presynaptic cytomatrix protein","acylaminoacyl-peptide hydrolase","inturned planar cell polarity protein","ring finger protein 123","heat shock protein family A (Hsp70) member 4 like","major facilitator superfamily domain containing 8","abhydrolase domain containing 18","MON1 homolog A, secretory trafficking associated","macrophage stimulating 1 receptor","RAD54 like 2","testis expressed 264, ER-phagy receptor","glutamate metabotropic receptor 2","dual specificity phosphatase 7","POC1 centriolar protein A","protein phosphatase, Mg2+/Mn2+ dependent 1M","WD repeat domain 82","chromosome 4 open reading frame 3","serine protease 12","N-deacetylase and N-sulfotransferase 3","high mobility group box 2","Sin3A associated protein 30","stimulator of chondrogenesis 1","mitotic arrest deficient 2 like 1","annexin A5","microtubule associated protein 9","guanylate cyclase 1 soluble subunit alpha 1","F-box protein 8","centrosomal protein 44","15-hydroxyprostaglandin dehydrogenase","transmembrane protein 144","golgi associated kinase 1B","N(alpha)-acetyltransferase 15, NatA auxiliary subunit","interleukin 15","family with sequence similarity 160 member A1","ADP ribosylation factor interacting protein 1","interactor of little elongation complex ELL subunit 1","hedgehog interacting protein","anaphase promoting complex subunit 10","ATP binding cassette subfamily E member 1","OTU deubiquitinase 4","LSM6 homolog, U6 small nuclear RNA and mRNA degradation associated","transmembrane protein 184C","protein arginine methyltransferase 9","integrin subunit alpha 2","molybdenum cofactor synthesis 2","EGF like repeats and discoidin domains 3","transmembrane protein 161B","ELOVL fatty acid elongase 7","NADH:ubiquinone oxidoreductase complex assembly factor 2","zinc finger protein 474","LMBR1 domain containing 2","RAN binding protein 3 like","NIPBL cohesin loading factor","ring finger protein 180","adhesion G protein-coupled receptor V1","solute carrier family 25 member 46","StAR related lipid transfer domain containing 4","protein geranylgeranyltransferase type I subunit beta","coagulation factor II thrombin receptor like 2","coiled-coil domain containing 112","ankyrin repeat domain 33B","carboxymethylenebutenolidase homolog","chromosome 5 open reading frame 63","proline rich coiled-coil 1","F2R like trypsin receptor 1","angiogenic factor with G-patch and FHA domains 1","WD repeat domain 41","PR/SET domain 9","NADH:ubiquinone oxidoreductase subunit S4","secretoglobin family 3A member 2","serine peptidase inhibitor, Kazal type 1","GrpE like 2, mitochondrial","cell division cycle 20B","arylsulfatase family member K","Rho related BTB domain containing 3","glutathione peroxidase 8 (putative)","tigger transposable element derived 6","serine incorporator 5","ectonucleotide pyrophosphatase/phosphodiesterase 6","cancer antigen 1","caspase 3","primase and DNA directed polymerase","endoplasmic reticulum aminopeptidase 1","endoplasmic reticulum aminopeptidase 2","cardiomyopathy associated 5","EGF like, fibronectin type III and laminin G domains","cilia and flagella associated protein 97","RPTOR independent companion of MTOR complex 2","terminal nucleotidyltransferase 2","EBF transcription factor 1","ankyrin repeat family A member 2","ubiquitin like domain containing CTD phosphatase 1","UTP15 small subunit processome component","toll like receptor 3","kallikrein B1","NSA2 ribosome biogenesis factor","G elongation factor mitochondrial 2","coiled-coil domain containing 127","forkhead box Q1","adhesion G protein-coupled receptor F2","acyl-CoA synthetase long chain family member 6","septin 8","shroom family member 1","growth differentiation factor 9","ubiquinol-cytochrome c reductase complex III subunit VII","liver enriched antimicrobial peptide 2","gap junction protein beta 7","solute carrier family 35 member A1","glutamate ionotropic receptor kainate type subunit 2","cyclic GMP-AMP synthase","fatty acid binding protein 7","taxilin beta","Cbp/p300 interacting transactivator with Glu/Asp rich carboxy-terminal domain 2","CREB3 regulatory factor","discoidin, CUB and LCCL domain containing 1","sideroflexin 1","sterile alpha motif domain containing 3","transmembrane protein 200A","decaprenyl diphosphate synthase subunit 2","syntaxin binding protein 5","interleukin 31 receptor A","retinoic acid early transcript 1E","diacylglycerol lipase beta","KIAA0895","serine/threonine kinase 17a","transformer 2 alpha homolog","polypeptide N-acetylgalactosaminyltransferase 10","SAP30 like","ribosomal protein S14","hyperpolarization activated cyclic nucleotide gated potassium channel 1","myozenin 3","component of oligomeric golgi complex 5","base methyltransferase of 25S rRNA 2 homolog","G protein-coupled receptor 85","SLU7 homolog, splicing factor","RP9 pre-mRNA splicing factor","PTTG1 regulator of sister chromatid separation, securin","calcium modulating ligand","BMP binding endothelial regulator","RELT like 2","potassium two pore domain channel subfamily K member 5","kinesin family member 6","zinc finger protein 12","solute carrier family 29 member 4","STEAP family member 1","cell division cycle associated 7 like","Sp8 transcription factor","meiosis regulator for oocyte development","KIAA1324 like","ubiquitin specific peptidase 49","synaptotagmin like 3","IQ motif and ubiquitin domain containing","hes related family bHLH transcription factor with YRPW motif 1","zinc finger protein 704","fatty acid binding protein 5","sonic hedgehog signaling molecule","T cell activation RhoGTPase activating protein","collagen type I alpha 2 chain","charged multivesicular body protein 4C","solute carrier family 13 member 4","brain protein I3","lemur tyrosine kinase 2","cathepsin B","DLC1 Rho GTPase activating protein","adenylate cyclase 1","chromosome 7 open reading frame 57","hepatocyte nuclear factor 4 gamma","peroxisomal biogenesis factor 2","RAD21 cohesin complex component","solute carrier family 30 member 8","mediator complex subunit 30","TNF receptor superfamily member 11b","somatomedin B and thrombospondin type 1 domain containing","phosphorylase kinase catalytic subunit gamma 1","potassium voltage-gated channel modifier subfamily V member 1","CUB and Sushi multiple domains 3","scaffold protein involved in DNA repair","origin recognition complex subunit 5","dynein axonemal assembly factor 5","oxidative stress induced growth inhibitor family member 2","defensin beta 1","Sad1 and UNC84 domain containing 1","oxidation resistance 1","G protein-coupled receptor 146","G protein-coupled estrogen receptor 1","transmembrane protein 184A","nitric oxide synthase 3","MICAL like 2","carbonic anhydrase 3","integrator complex subunit 1","cyclin dependent kinase 5","solute carrier family 4 member 2","Fas activated serine/threonine kinase","transmembrane and ubiquitin like domain containing 1","formation of mitochondrial complex V assembly factor 1 homolog","phosphorylated adaptor for RNA export","aldehyde dehydrogenase 7 family member A1","forkhead box K1","cytochrome c oxidase subunit 6C","tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta","BAALC binder of MAP3K1 and KLF4","frizzled class receptor 6","solute carrier family 25 member 32","DDB1 and CUL4 associated factor 13","tumor protein p53 inducible nuclear protein 1","integrator complex subunit 8","vir like m6A methyltransferase associated","GTP binding protein overexpressed in skeletal muscle","pyruvate dehyrogenase phosphatase catalytic subunit 1","transmembrane protein 67","WASH complex subunit 5","ribonuclease P/MRP subunit p25 like","family with sequence similarity 219 member A","chromosome 9 open reading frame 24","small nuclear RNA activating complex polypeptide 3","myogenesis regulating glycosidase (putative)","nudix hydrolase 2","transmembrane protein 65","PC4 and SFRS1 interacting protein 1","coiled-coil domain containing 171","ubiquitin associated protein 1","spleen associated tyrosine kinase","nipsnap homolog 3B","ATP binding cassette subfamily A member 1","nuclear factor, interleukin 3 regulated","leucine zipper and EF-hand containing transmembrane protein 2","methyltransferase like 2B","frataxin","zinc finger matrin-type 4","transmembrane protein 71","MAM domain containing 2","chromosome 8 open reading frame 34","transmembrane channel like 1","aldehyde dehydrogenase 1 family member A1","lysine demethylase 1B","heparan-alpha-glucosaminide N-acetyltransferase","RAS and EF-hand domain containing","G kinase anchoring protein 1","kinesin family member 27","chromosome 9 open reading frame 64","heterogeneous nuclear ribonucleoprotein K","sushi, von Willebrand factor type A, EGF and pentraxin domain containing 1","transient receptor potential cation channel subfamily V member 6","ankyrin repeat and sterile alpha motif domain containing 6","fructose-bisphosphatase 1","post-GPI attachment to proteins GalNAc transferase 4","zinc fingers and homeoboxes 1","cilia and flagella associated protein 47","cytochrome b-245 beta chain","dynein light chain Tctex-type 3","methyltransferase like 27","MID1 interacting protein 1","shortage in chiasmata 1","chromosome X open reading frame 58","KIAA1958","patched domain containing 1","ring finger protein 183","protocadherin 19","phosphatidylinositol glycan anchor biosynthesis class A","spermatid perinuclear RNA binding protein","claudin 3","GTPase activating protein and VPS9 domains 1","caspase recruitment domain family member 19","WNK lysine deficient protein kinase 2","ATPase copper transporting alpha","zinc finger protein 367","neuroligin 4 Y-linked","highly divergent homeobox","NADH:ubiquinone oxidoreductase subunit B6","aquaporin 7","nucleolar protein 6","aquaporin 3 (Gill blood group)","tRNA methyltransferase 10B","valosin containing protein","phosphatidylinositol glycan anchor biosynthesis class O","stomatin like 2","bromodomain and WD repeat domain containing 3","SLIT and NTRK like family member 5","maternal embryonic leucine zipper kinase","armadillo repeat containing 3","OTU deubiquitinase 1","Rho GTPase activating protein 12","FAT atypical cadherin 3","deuterosome assembly protein 1","HECT domain E3 ubiquitin protein ligase 2","F-box protein 33","integrator complex subunit 6 like","leucine rich repeat containing 18","serine palmitoyltransferase small subunit A","WRN RecQ like helicase","membrane associated ring-CH-type finger 8","thyroid stimulating hormone receptor","cofilin 2","SGT1 homolog, MIS12 kinetochore complex assembly cochaperone","general transcription factor IIA subunit 1","zinc finger CCHC-type containing 24","phosphoglucomutase 2 like 1","phytanoyl-CoA 2-hydroxylase interacting protein like","solute carrier family 16 member 9","inositol polyphosphate phosphatase like 1","gap junction protein beta 2","crystallin lambda 1","receptor accessory protein 3","hepatic and glial cell adhesion molecule","spindle and kinetochore associated complex subunit 3","mitochondrial calcium uptake 2","DNA damage induced apoptosis suppressor","PCF11 cleavage and polyadenylation factor subunit","leucine rich repeat protein 1","ribosomal protein L36a like","dynein axonemal assembly factor 2","DEPP1 autophagy regulator","zinc finger protein 22","kelch domain containing 2","EMAP like 5","nuclear export mediator factor","RNA pseudouridine synthase D4","ADP ribosylation factor 6","tetratricopeptide repeat domain 8","transmembrane protein 63C","NADP dependent oxidoreductase domain containing 1","APC membrane recruitment protein 2","aldo-keto reductase family 1 member E2","kelch repeat and BTB domain containing 6","orthodenticle homeobox 2","fatty acid amide hydrolase 2","nudix hydrolase 5","BEN domain containing 7","ATP synthase F1 subunit gamma","pre-mRNA processing factor 18","TATA-box binding protein associated factor 3","V-set and transmembrane domain containing 4","voltage dependent anion channel 2","catechol-O-methyltransferase domain containing 1","solute carrier family 18 member A2","PDZ domain containing 8","zinc finger protein 503","abraxas 2, BRISC complex subunit","quiescin sulfhydryl oxidase 2","family with sequence similarity 204 member A","nuclear receptor binding SET domain protein 1","peroxiredoxin 3","ecto-NOX disulfide-thiol exchanger 2","growth hormone inducible transmembrane protein","C-type lectin domain family 1 member B","small nuclear RNA activating complex polypeptide 4","transmembrane protein 52B","peptidase, mitochondrial processing alpha subunit","endosome associated trafficking regulator 1","adenylate kinase 8","sperm acrosome associated 9","TSC complex subunit 1","growth factor independent 1B transcriptional repressor","hypoxanthine phosphoribosyltransferase 1","BLOC-1 related complex subunit 5","divergent protein kinase domain 1B","zinc finger MYND-type containing 19","storkhead box 1","ret proto-oncogene","DExD-box helicase 21","BMS1 ribosome biogenesis factor","serine/threonine kinase 32C","junctional cadherin 5 associated","olfactory receptor family 4 subfamily K member 2","FUN14 domain containing 2","phosphatidylinositol-4,5-bisphosphate 4-phosphatase 1","methyltransferase like 17","NDRG family member 2","ribonuclease A family member 7","Rho guanine nucleotide exchange factor 40","NMDA receptor synaptonuclear signaling and neuronal migration factor","zinc finger protein 219","chromosome 12 open reading frame 50","caspase 7","protein phosphatase 1 regulatory subunit 36","butyrophilin like 9","coiled-coil domain containing 186","von Willebrand factor A domain containing 2","methyltransferase like 3","spalt like transcription factor 2","TruB pseudouridine synthase family member 1","cytochrome P450 family 2 subfamily C member 19","zinc finger FYVE-type containing 1","chromosome 10 open reading frame 82","heat shock protein family A (Hsp70) member 12A","FRAT regulator of WNT signaling pathway 1","ubiquitin domain containing 1","E2F transcription factor 7","Rho GTPase activating protein 42","iron-sulfur cluster assembly 2","LARGE xylosyl- and glucuronyltransferase 2","protein kinase C and casein kinase substrate in neurons 3","tetratricopeptide repeat domain 7B","solute carrier family 39 member 13","proteasome 26S subunit, ATPase 3","receptor associated protein of the synapse","ATP/GTP binding protein like 2","tandem C2 domains, nuclear","cleavage and polyadenylation specific factor 2","modulator of apoptosis 1","interferon alpha inducible protein 27 like 1","interferon alpha inducible protein 27","calmin","coiled-coil domain containing 38","phosphotriesterase related","complement C1q like 3","calcium voltage-gated channel auxiliary subunit beta 2","3-hydroxyacyl-CoA dehydratase 1","ADP ribosylation factor like GTPase 5B","single-pass membrane protein with coiled-coil domains 4","centrosomal protein 295","TATA-box binding protein associated factor, RNA polymerase I subunit D","ankyrin repeat and BTB domain containing 2","R3H domain and coiled-coil containing 1 like","angiomotin like 1","HtrA serine peptidase 1","lipase C, hepatic type","centrosomal protein 57","t-complex 11 like 2","sprouty related EVH1 domain containing 1","transmembrane and coiled-coil domains 5A","junctional adhesion molecule 3","galactosidase beta 1 like 3","SV2 related protein","glutamic--pyruvic transaminase 2","amnion associated transmembrane protein","RAB8B, member RAS oncogene family","IKBKB interacting protein","RNA pseudouridine synthase domain containing 2","hypoxia inducible factor 1 subunit alpha inhibitor","NADH:ubiquinone oxidoreductase subunit B8","zinc finger FYVE-type containing 19","serine peptidase inhibitor, Kunitz type 1","fibrillin 1","DEP domain containing 4","bromodomain containing 7","creatine kinase B","tRNA methyltransferase 61A","beta-transducin repeat containing E3 ubiquitin protein ligase","DNA polymerase lambda","BCL2 associated athanogene 5","deleted in primary ciliary dyskinesia homolog (mouse)","La ribonucleoprotein 6, translational regulator","apoptosis inhibitor 5","asparaginase","zinc finger protein 319","HPS6 biogenesis of lysosomal organelles complex 2 subunit 3","SUMO peptidase family member, NEDD8 specific","nucleolar and coiled-body phosphoprotein 1","alkB homolog 3, alpha-ketoglutaratedependent dioxygenase","COP9 signalosome subunit 2","gamma-aminobutyric acid type A receptor beta3 subunit","sphingosine-1-phosphate lyase 1","fibroblast growth factor receptor substrate 2","chaperonin containing TCP1 subunit 2","pterin-4 alpha-carbinolamine dehydratase 1","ariadne RBR E3 ubiquitin protein ligase 1","chromosome 16 open reading frame 71","CXADR like membrane protein","sodium voltage-gated channel beta subunit 3","cytochrome c oxidase copper chaperone COX11","zinc finger protein 202","family with sequence similarity 227 member B","syntaxin binding protein 4","cysteine and tyrosine rich 1","cullin 5","myelin regulatory factor like","WW domain binding protein 1 like","BLOC-1 related complex subunit 7","complement C2","pleckstrin homology and FYVE domain containing 1","anaphase promoting complex subunit 16","sphingomyelin phosphodiesterase 1","amyloid beta precursor protein binding family B member 1","synaptopodin 2 like","nudix hydrolase 13","chromosome 11 open reading frame 65","tripartite motif containing 44","integrin linked kinase","TATA-box binding protein associated factor 10","tripeptidyl peptidase 1","dachsous cadherin-related 1","MSS51 mitochondrial translational activator","cytochrome b5 type A","ubiquitin specific peptidase 54","recombination activating 1","chromosome 11 open reading frame 74","WD repeat domain 88","ATPase phospholipid transporting 9B (putative)","PPFIA binding protein 2","cytochrome b5 reductase 2","serpin family B member 7","KIAA0355","serpin family B member 8","TUB bipartite transcription factor","RIC3 acetylcholine receptor chaperone","isocitrate dehydrogenase (NAD(+)) 3 catalytic subunit alpha","WD repeat domain 72","phospholipase D family member 4","zinc finger matrin-type 1","X-ray radiation resistance associated 1","tripartite motif containing 66","ring finger protein 169","ribosomal protein L27a","DENN domain containing 2B","chromodomain Y like 2","transmembrane protein 130","centromere protein N","A-kinase interacting protein 1","ATM interactor","chromosome 16 open reading frame 46","transmembrane protein 41B","LEO1 homolog, Paf1/RNA polymerase II complex component","zinc finger protein 143","thioredoxin related transmembrane protein 3","WEE1 G2 checkpoint kinase","mitogen-activated protein kinase 7","protein kinase C beta","HDGF like 3","N-deacetylase and N-sulfotransferase 2","minichromosome maintenance complex component 7","C-type lectin domain family 3 member A","coiled-coil domain containing 68","C-type lectin domain family 4 member E","zinc finger protein 3","C-type lectin domain family 4 member D","zinc finger and SCAN domain containing 21","ribosomal modification protein rimK like family member B","alpha-2-macroglobulin like 1","brain expressed associated with NEDD4 1","thymidine kinase 2","transmembrane p24 trafficking protein 3","SEC11 homolog C, signal peptidase complex subunit","complexin 4","galanin receptor 1","transmembrane protein 135","IQ motif containing D","nudE neurodevelopment protein 1 like 1","centromere protein V","cadherin 16","RRAD, Ras related glycolysis inhibitor and calcium channel regulator","cytosolic iron-sulfur assembly component 2B","cilia and flagella associated protein 52","heat shock protein 90 beta family member 1","BLCAP apoptosis inducing factor","CHRNA7 (exons 5-10) and FAM7A (exons A-E) fusion","activating transcription factor 7 interacting protein 2","matrix metallopeptidase 10","trans-golgi network vesicle protein 23 homolog A","brain expressed X-linked 3","transmembrane serine protease 5","component of oligomeric golgi complex 1","pleckstrin homology domain containing A7","zinc finger protein 606","beta-2-microglobulin","zinc finger protein 592","cancer susceptibility 4","nicotinamide N-methyltransferase","acyl-CoA synthetase medium chain family member 1","adaptor related protein complex 1 subunit gamma 1","schlafen family member 5","cation channel sperm associated 2","bMERB domain containing 1","meiosis regulator and mRNA stability factor 1","serum amyloid A like 1","peptidylprolyl isomerase B","lactate dehydrogenase C","cytosolic iron-sulfur assembly component 2A","lactate dehydrogenase A like 6A","family with sequence similarity 111 member A","PCNA clamp associated factor","kinesin family member 7","lactate dehydrogenase D","perilipin 1","peroxisomal biogenesis factor 11 alpha","transmembrane protein 170A","mesoderm posterior bHLH transcription factor 1","alanyl aminopeptidase, membrane","sodium channel epithelial 1 gamma subunit","neuron navigator 2","ankyrin repeat and death domain containing 1A","glycine-N-acyltransferase like 1","chromosome 18 open reading frame 54","dynactin subunit 5","TERF2 interacting protein","polo like kinase 1","caseinolytic mitochondrial matrix peptidase chaperone subunit","zinc finger and BTB domain containing 39","calcium voltage-gated channel auxiliary subunit gamma 2","myosin IA","calcineurin like EF-hand protein 2","nuclear envelope integral membrane protein 1","NGFI-A binding protein 2","VPS39 subunit of HOPS complex","signal transducer and activator of transcription 6","PAT1 homolog 1, processing body mRNA decay factor","ATP23 metallopeptidase and ATP synthase assembly factor homolog","syntaxin 3","mitochondrial ribosomal protein L16","phosphatidylinositol-5-phosphate 4-kinase type 2 gamma","myotubularin related protein 10","tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein beta","chromosome 15 open reading frame 48","gremlin 1, DAN family BMP antagonist","neuronal tyrosine phosphorylated phosphoinositide-3-kinase adaptor 1","TSC22 domain family member 4","membrane spanning 4-domains A7","membrane spanning 4-domains A14","DIS3 like exosome 3'-5' exoribonuclease","cyclin D1 binding protein 1","erythrocyte membrane protein band 4.2","SMAD family member 3","membrane spanning 4-domains A8","coiled-coil domain containing 178","microtubule associated protein 1A","RCC1 domain containing 1","AKT interacting protein","microtubule associated protein RP/EB family member 2","eva-1 homolog C","methionyl-tRNA synthetase 1","methyl-CpG binding domain protein 6","canopy FGF signaling regulator 4","protein disulfide isomerase family A member 3","nudix hydrolase 21","N-acetyltransferase 16 (putative)","telomere repeat binding bouquet formation protein 2","NK3 homeobox 1","small G protein signaling modulator 1","dual specificity phosphatase 18","TEF transcription factor, PAR bZIP family member","meiotic double-stranded break formation protein 1","beta-1,4-N-acetyl-galactosaminyltransferase 2","PBX homeobox 3","G protein subunit gamma transducin 2","prohibitin","small nuclear ribonucleoprotein D1 polypeptide","tetratricopeptide repeat domain 16","sterile alpha motif domain containing 14","phosphatidylinositol-4-phosphate 5-kinase like 1","transmembrane protein 92","family with sequence similarity 102 member A","acyl-CoA synthetase family member 2","golgin A2","TruB pseudouridine synthase family member 2","coenzyme Q4","solute carrier family 27 member 4","ANKRD40 C-terminal like","ubiquitin related modifier 1","cerebral endothelial cell adhesion molecule","dolichyldiphosphatase 1","coiled-coil domain containing 103","endonuclease G","paired related homeobox 2","UDP glucuronosyltransferase family 1 member A6","chromosome 15 open reading frame 39","immunoglobulin superfamily containing leucine rich repeat 2","Sp2 transcription factor","proline rich 15 like","coenzyme Q7, hydroxylase","G protein-coupled receptor class C group 5 member B","CRK proto-oncogene, adaptor protein","F-box protein 22","TBC1 domain family member 2B","nucleotide binding oligomerization domain containing 2","sorting nexin 20","lipoxygenase homology domains 1","katanin catalytic subunit A1 like 2","haloacid dehalogenase like hydrolase domain containing 2","zinc finger protein 91","insulin like growth factor 2","ring finger protein 214","cyclin dependent kinase 12","dipeptidase 2","dihydrouridine synthase 2","POP5 homolog, ribonuclease P/MRP subunit","endo-beta-N-acetylglucosaminidase","RNA binding fox-1 homolog 3","ATP synthase membrane subunit g","CD3d molecule","TBC1 domain family member 16","TEPSIN adaptor related protein complex 4 accessory protein","myosin VB","acetyl-CoA acyltransferase 2","stromal interaction molecule 1","ribonucleotide reductase catalytic subunit M1","olfactory receptor family 51 subfamily E member 2","tripartite motif containing 68","olfactory receptor family 51 subfamily B member 5","fructosamine 3 kinase","proline rich transmembrane protein 2","zinc finger protein 23","immunity related GTPase Q","zinc finger protein 226","zinc finger protein 180","protein phosphatase 2 regulatory subunit B''beta","zinc finger protein 668","zinc finger protein 646","vitamin K epoxide reductase complex subunit 1","lactoperoxidase","carbonic anhydrase 4","SMG8 nonsense mediated mRNA decay factor","tropomyosin 4","RAB8A, member RAS oncogene family","glutathione peroxidase 4","midnolin","junctional sarcoplasmic reticulum protein 1","niban apoptosis regulator 3","kelch like family member 26","GATA zinc finger domain containing 2A","mevalonate diphosphate decarboxylase","chromatin licensing and DNA replication factor 1","trafficking protein particle complex 2 like","ankyrin repeat domain 11","spermatogenesis associated 33","ribosomal protein S6 kinase related","protein interacting with cyclin A1","ribosomal protein L13","zinc finger protein 641","calcium voltage-gated channel auxiliary subunit beta 3","dehydrogenase/reductase 13","tumor protein p53 inducible protein 13","lysine methyltransferase 2D","coronin 6","RHEB like 1","tubulin alpha 1a","tubulin alpha 1c","zinc finger protein 610","zinc finger protein 528","zinc finger protein 701","SERTA domain containing 3","NCK associated protein 5 like","RAB4B, member RAS oncogene family","aquaporin 2","glycerol-3-phosphate dehydrogenase 1","proline and serine rich 3","cytochrome P450 family 2 subfamily S member 1","AXL receptor tyrosine kinase","NFKB inhibitor delta","transmembrane channel like 4","leukocyte associated immunoglobulin like receptor 1","tweety family member 1","leukocyte receptor cluster member 8","CDC42 effector protein 5","transmembrane protein 145","zinc finger protein 526","trafficking protein particle complex 9","zinc finger protein 146","zinc finger protein 283","serine peptidase inhibitor, Kunitz type 2","chromosome 19 open reading frame 33","Yip1 interacting factor homolog B, membrane trafficking protein","dynein axonemal assembly factor 3","prostate stem cell antigen","ATCAY kinesin light chain interacting caytaxin","lymphocyte antigen 6 family member D","death associated protein kinase 3","eukaryotic translation elongation factor 2","transmembrane and immunoglobulin domain containing 2","chromatin assembly factor 1 subunit A","UBX domain protein 6","HDGF like 2","perilipin 4","semaphorin 6B","zinc finger protein 444","nucleoredoxin","TLC domain containing 3A","glyoxalase domain containing 4","major facilitator superfamily domain containing 3","glutamic--pyruvic transaminase","kinesin family member C2","solute carrier family 43 member 2","Rab interacting lysosomal protein","serpin family F member 2","WD repeat domain 81","serine racemase","TSR1 ribosome maturation factor","transient receptor potential cation channel subfamily V member 3","hydroxysteroid 11-beta dehydrogenase 1 like","cytochrome b5 domain containing 2","gamma-glutamyltransferase 6","chromosome 19 open reading frame 48","kallikrein related peptidase 4","kallikrein related peptidase 2","kallikrein related peptidase 6","kallikrein related peptidase 11","kallikrein related peptidase 13","zinc finger protein 83","keratin 80","OTU deubiquitinase, ubiquitin aldehyde binding 1","REST corepressor 2","angiopoietin like 4","CD320 molecule","SPRY domain containing 3","zinc finger protein 558","NADH:ubiquinone oxidoreductase core subunit V1","cyclin dependent kinase 2 associated protein 2","nudix hydrolase 8",null,"peroxiredoxin 2","zinc finger protein 232","MIS12 kinetochore complex component","CD300a molecule","tektin 1","HID1 domain containing","mitochondrial ribosomal protein L58","ATP synthase peripheral stalk subunit d","transmembrane protein 88","envoplakin","signal recognition particle 68","transmembrane channel like 8","thymidine kinase 1","transmembrane protein 68","gasdermin A","keratin 24","GH3 domain containing","family with sequence similarity 234 member A","zinc finger protein 598","RAB26, member RAS oncogene family","MTOR associated protein, LST8 homolog","E4F transcription factor 1","deoxyribonuclease 1 like 2","enoyl-CoA delta isomerase 1","CASK interacting protein 1","ATP binding cassette subfamily A member 3","potassium channel tetramerization domain containing 5","serine/arginine repetitive matrix 2","zinc finger protein 597","NLR family CARD domain containing 3","succinate dehydrogenase complex assembly factor 2","damage specific DNA binding protein 1","VPS37C subunit of ESCRT-I","von Willebrand factor C and EGF domains","RAB3A interacting protein like 1","bestrophin 1","ferritin heavy chain 1","BSCL2 lipid droplet biogenesis associated, seipin","RNA polymerase II subunit G","solute carrier family 3 member 2","spindlin interactor and repressor of chromatin binding","autophagy related 16 like 2","C2 domain containing 3 centriole elongation regulator","tetratricopeptide repeat and ankyrin repeat containing 1","tetratricopeptide repeat domain 21A","ribosomal protein SA","ectonucleoside triphosphate diphosphohydrolase 3","catenin beta 1","unc-51 like kinase 4","Fas associated via death domain","latent transforming growth factor beta binding protein 3","SAC3 domain containing 1","basic leucine zipper ATF-like transcription factor 2","solute carrier family 22 member 11","splicing factor 1","mitogen-activated protein kinase kinase kinase kinase 2","coiled-coil domain containing 88B","scavenger receptor class A member 3","prepronociceptin","COP9 signalosome subunit 6","platelet activating factor acetylhydrolase 1b catalytic subunit 2","ankyrin repeat and sterile alpha motif domain containing 3","nudix hydrolase 16 like 1","KIAA1586","RAB4A, member RAS oncogene family","SET domain containing 5","vasorin","family with sequence similarity 83 member B","THAP domain containing 9","ring finger protein 187","hook microtubule tethering protein 3","mitogen-activated protein kinase 1 interacting protein 1 like","DNA damage inducible transcript 4","recombination signal binding protein for immunoglobulin kappa J region","LMBR1 domain containing 1","zinc finger CCHC-type containing 4","prostaglandin D2 receptor","tetratricopeptide repeat domain 39C","glycerate kinase","G protein subunit gamma 4","ubiquitin domain containing 2","RNA polymerase II subunit J3","NFKB inhibitor interacting Ras like 2","DnaJ heat shock protein family (Hsp40) member C7","interferon regulatory factor 2 binding protein 2","5'-nucleotidase domain containing 2","forkhead box I1","small integral membrane protein 4","cytochrome c oxidase assembly factor 6","kinesin family member 5C","alpha-1,6-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase","BMI1 proto-oncogene, polycomb ring finger","THAP domain containing 11","metabolism of cobalamin associated D","pyruvate dehydrogenase E1 beta subunit","PX domain containing serine/threonine kinase like","H1.4 linker histone, cluster member","protein-L-isoaspartate (D-aspartate) O-methyltransferase domain containing 1","potassium channel tetramerization domain containing 6","M-phase specific PLK1 interacting protein","acyl-CoA oxidase 2","family with sequence similarity 107 member A","interferon regulatory factor 2","myelin associated oligodendrocyte basic protein","C-X3-C motif chemokine receptor 1","xin actin binding repeat containing 1","delta 4-desaturase, sphingolipid 2","sodium voltage-gated channel alpha subunit 11","ADP ribosylation factor 4","septin 2","filamin A interacting protein 1 like","major facilitator superfamily domain containing 2A","deoxythymidylate kinase","transporter 1, ATP binding cassette subfamily B member","inhibitor of growth family member 5","autophagy related 4B cysteine peptidase","bradykinin receptor B2","mixed lineage kinase domain like pseudokinase","ring finger and WD repeat domain 3","melatonin receptor 1A","potassium voltage-gated channel modifier subfamily G member 4","ras homolog family member H","component of oligomeric golgi complex 7","cell division cycle 40","stress induced phosphoprotein 1","sodium channel epithelial 1 beta subunit","HR lysine demethylase and nuclear receptor corepressor","thioredoxin domain containing 2","RAB31, member RAS oncogene family","receptor accessory protein 4","tenascin XB","bone morphogenetic protein 1","ataxin 2 like","coiled-coil domain containing 110","RNA polymerase III subunit D","flap structure-specific endonuclease 1","caveolae associated protein 2","microtubule crosslinking factor 1","HEXIM P-TEFb complex subunit 2","farnesyltransferase, CAAX box, alpha","serine incorporator 2","trafficking protein particle complex 11","inhibitor of growth family member 2","CDKN2A interacting protein","small nuclear ribonucleoprotein U11/U12 subunit 48","transmembrane protein 223","solute carrier family 20 member 2","dynein light chain roadblock-type 2","transmembrane and ubiquitin like domain containing 2","ADAM metallopeptidase domain 29","signal transducer and activator of transcription 3","zinc finger SWIM-type containing 1","ADAM metallopeptidase domain 9","glial cell derived neurotrophic factor","axin 2","NADH:ubiquinone oxidoreductase subunit S5","von Willebrand factor A domain containing 3B","zinc finger protein 30","LRAT domain containing 2","low density lipoprotein receptor class A domain containing 4","potassium channel tetramerization domain containing 19","solute carrier family 16 member 4","interleukin 7 receptor","transmembrane protein 208","LDL receptor related protein 1B","WAP four-disulfide core domain 12","adenosylhomocysteinase like 1","DnaJ heat shock protein family (Hsp40) member C21","cAMP-dependent protein kinase inhibitor gamma","nephronectin","family with sequence similarity 178 member B","semaphorin 4C","cyclin and CBS domain divalent metal cation transport mediator 3","glutathione S-transferase mu 4","tet methylcytosine dioxygenase 2","CXXC finger protein 4","tectonic family member 2","diphosphoinositol pentakisphosphate kinase 1","tetraspanin 5","abhydrolase domain containing 15","zinc finger and BTB domain containing 5","chromosome transmission fidelity factor 8","adenosine deaminase like","leucine carboxyl methyltransferase 2","syntrophin beta 2","interleukin 12A","zinc finger protein 507","syntaxin 18","neuronal vesicle trafficking associated 1","zinc finger and BTB domain containing 49","G elongation factor mitochondrial 1","DEAD-box helicase 19A","atonal bHLH transcription factor 8","ankyrin repeat domain 49","surfactant protein B","ubiquitin specific peptidase 39","TNFAIP3 interacting protein 2","chromosome 2 open reading frame 68","transmembrane protein 150A","ring finger protein 181","vesicle associated membrane protein 5","leucine rich repeat containing 28","methionine adenosyltransferase 2A","phospholipase A2 group IVF","energy homeostasis associated","zinc finger protein 608","solute carrier family 35 member G2","inositol polyphosphate-5-phosphatase D","leucine zipper and EF-hand containing transmembrane protein 1","transmembrane protein 129","peptidylprolyl isomerase C","sprouty RTK signaling antagonist 3","centrosomal protein 120","syntaxin binding protein 6","transmembrane 4 L six family member 20","mitochondrial fission factor","glutamate metabotropic receptor 5","galectin 9","JMJD7-PLA2G4B readthrough","PX domain containing 1","sialic acid binding Ig like lectin 7","E2F transcription factor 6","fem-1 homolog B","COMM domain containing 8","ATP synthase membrane subunit e","ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1","solute carrier family 49 member 3","collagen type IV alpha 3 chain","mitogen-activated protein kinase kinase 1","kallikrein related peptidase 7","heterogeneous nuclear ribonucleoprotein H1","insulin receptor substrate 1","methyl-CpG binding protein 2","UPF3A regulator of nonsense mediated mRNA decay","zinc finger B-box domain containing","androgen receptor","dehydrogenase/reductase X-linked","vexin","HSPB1 associated protein 1","acetylserotonin O-methyltransferase like","solute carrier family 25 member 6","carbohydrate sulfotransferase 14","prostate androgen-regulated mucin-like protein 1","casein kinase 1 gamma 1","family with sequence similarity 110 member B","armadillo repeat containing 4","actin filament associated protein 1 like 2","zinc finger protein 354A","activating transcription factor 5","ubiquitin conjugating enzyme E2 V2","zinc finger and BTB domain containing 43","carnitine palmitoyltransferase 1C","proprotein convertase subtilisin/kexin type 9","exportin 6","GSG1 like","MN1 proto-oncogene, transcriptional regulator","apurinic/apyrimidinic endodeoxyribonuclease 2","NSE1 homolog, SMC5-SMC6 complex component","coiled-coil domain containing 126","nuclear pore complex interacting protein family member B12","olfactory receptor family 10 subfamily G member 3","RAB3B, member RAS oncogene family","CD2 cytoplasmic tail binding protein 2","regulator of G protein signaling 14","TBC1 domain family member 10B","lectin, mannose binding 2","germinal center associated signaling and motility like","RAB24, member RAS oncogene family","PRELI domain containing 1","thrombospondin 3","carbonic anhydrase 5B","solute carrier family 50 member 1","ephrin A1","C-X-C motif chemokine ligand 10","nuclear pore complex interacting protein family member B3","SH3 domain and tetratricopeptide repeats 2","C-X-C motif chemokine ligand 11","zinc finger CCCH-type, RNA binding motif and serine/arginine rich 2","NMD3 ribosome export adaptor","adrenoceptor beta 2","beta-1,3-N-acetylgalactosaminyltransferase 1 (globoside blood group)","G protein regulated inducer of neurite outgrowth 1","potassium voltage-gated channel subfamily A member regulatory beta subunit 1","mitochondrial ribosomal protein L1","Src homology 2 domain containing E","phosphoglucomutase 2","serine/threonine kinase 32A","chromosome 22 open reading frame 15","olfactory receptor family 5 subfamily AU member 1","membrane integral NOTCH2 associated receptor 1","glycoprotein 2","solute carrier family 33 member 1","snurportin 1","CASP2 and RIPK1 domain containing adaptor with death domain","SIN3 transcription regulator family member A","ADP ribosylation factor like GTPase 13B","protein tyrosine kinase 2","radial spoke head 10 homolog B2","platelet activating factor receptor","protein tyrosine phosphatase non-receptor type 9","ribonuclease A family member k6","C-X-C motif chemokine ligand 8","sodium voltage-gated channel alpha subunit 9","Ras association domain family member 6","syndecan 2","CD52 molecule","membrane magnesium transporter 1","small proline rich protein 1B","small proline rich protein 1A","TM2 domain containing 2","HtrA serine peptidase 4","pleckstrin homology domain containing A2","chloride intracellular channel 4","solute carrier family 38 member 11","G protein-coupled receptor 183","cysteine rich C-terminal 1","coiled-coil domain containing 8","methyltransferase like 15","mucin 15, cell surface associated","zinc finger E-box binding homeobox 2","gap junction protein beta 1","poly(rC) binding protein 1","histidine triad nucleotide binding protein 1","DTW domain containing 2","chloride intracellular channel 3","INO80 complex subunit E","basonuclin 1","DNA fragmentation factor subunit beta","NFU1 iron-sulfur cluster scaffold","ANTXR cell adhesion molecule 1","cytoskeleton associated protein 2 like","chromosome 15 open reading frame 40","RNA guanine-7 methyltransferase activating subunit","aprataxin and PNKP like factor","bolA family member 2B","RANBP2 like and GRIP domain containing 8","HIC ZBTB transcriptional repressor 2","leucine zipper protein 1","hexosaminidase D","BUB1 mitotic checkpoint serine/threonine kinase","sphingolipid transporter 1 (putative)","leucine rich repeat containing 45","cholinergic receptor nicotinic alpha 5 subunit","centromere protein X","1-acylglycerol-3-phosphate O-acyltransferase 2","ASPSCR1 tether for SLC2A4, UBX domain containing","fatty acid synthase","CCHC-type zinc finger nucleic acid binding protein","metallothionein 1E","dihydrouridine synthase 1 like","G protein pathway suppressor 1","RFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase","dicarbonyl and L-xylulose reductase","zinc finger protein 32","LIM domain binding 2","Rac family small GTPase 3","neuregulin 4","LIM zinc finger domain containing 1","transmembrane protein 266","transmembrane anterior posterior transformation 1","UDP-glucose pyrophosphorylase 2","heterogeneous nuclear ribonucleoprotein F","biotinidase","chondroitin sulfate N-acetylgalactosaminyltransferase 2","protocadherin 7","roundabout guidance receptor 1","one cut homeobox 1","apoptosis and caspase activation inhibitor","purinergic receptor P2Y1","tripartite motif containing 56","mucin 17, cell surface associated","RALBP1 associated Eps domain containing 2","mucin 3A, cell surface associated","synapse associated protein 1","integrin subunit alpha M","pyrin domain containing 1","tyrosylprotein sulfotransferase 1","torsin 1A interacting protein 2","transmembrane 4 L six family member 1","OTU deubiquitinase 3","OTU deubiquitinase 7A","glucuronidase beta","bromodomain containing 3","Kruppel like factor 13","zinc finger protein, FOG family member 2","zinc finger protein 764","zinc finger protein 747","zinc finger protein 768","taste 1 receptor member 3","transmembrane protein 42","mitogen-activated protein kinase kinase kinase 2","pseudouridine synthase like 1","splicing factor 3b subunit 5","zinc finger protein 35","tigger transposable element derived 4","intermediate filament family orphan 2","neuroligin 2","myosin VIIB","chromodomain helicase DNA binding protein 3","transmembrane protein 154","myosin VIIA and Rab interacting protein","activated leukocyte cell adhesion molecule","tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein gamma","ubiquitin conjugating enzyme E2 E3","centrobin, centriole duplication and spindle assembly protein","trafficking protein particle complex 1","potassium voltage-gated channel subfamily A regulatory beta subunit 3","family with sequence similarity 153 member A","G protein-coupled receptor 37 like 1","SUMO interacting motifs containing 1","transmembrane protein 192","speedy/RINGO cell cycle regulator family member E5","zinc finger protein 778","NIPA magnesium transporter 1","G protein-coupled receptor 25","ubiquitin conjugating enzyme E2 E1","heterogeneous nuclear ribonucleoprotein A3","salt inducible kinase 2","ring finger protein 150","coiled-coil domain containing 144A","cholinergic receptor nicotinic beta 1 subunit","glycophorin A (MNS blood group)","ubiquitin specific peptidase 38","solute carrier family 16 member 5","N-acetylneuraminic acid phosphatase","ankyrin repeat and kinase domain containing 1","ADP-ribose/CDP-alcohol diphosphatase, manganese dependent","fatty acid binding protein 6","PWWP domain containing 2A","ubiquitin specific peptidase 50","ubiquitin specific peptidase 47","programmed cell death 6 interacting protein","zinc finger protein 212","FAM161 centrosomal protein A","zinc finger protein 282","galactosidase beta 1","GON7 subunit of KEOPS complex","fatty acid hydroxylase domain containing 2","cartilage associated protein","cyclic nucleotide gated channel subunit beta 3","elongator acetyltransferase complex subunit 5","CKLF like MARVEL transmembrane domain containing 8","GABA type A receptor-associated protein","galectin 9B","syntaxin 8","cyclin dependent kinase 1","ubiquitin B","nuclear factor related to kappaB binding protein","FERM and PDZ domain containing 2","PR/SET domain 10","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 2","Fos proto-oncogene, AP-1 transcription factor subunit","transmembrane p24 trafficking protein 10","SET domain and mariner transposase fusion gene","SMAD family member 1","empty spiracles homeobox 2","TRPM8 channel associated factor 2","semaphorin 3E","leucine rich repeat neuronal 2","solute carrier family 30 member 1","doublecortin like kinase 2","G protein-coupled receptor class C group 5 member C","transmembrane protein 182","V-set and transmembrane domain containing 2A","keratin 8","keratin 78","adenosine A2b receptor","O-6-methylguanine-DNA methyltransferase","keratin 86","histidyl-tRNA synthetase 1","nuclear transcription factor, X-box binding like 1","DENN domain containing 5B","CD14 molecule","DnaJ heat shock protein family (Hsp40) member C18","ribosomal oxygenase 1","spermatogenesis associated 24","Ral GTPase activating protein non-catalytic beta subunit","PYM homolog 1, exon junction complex associated factor","marginal zone B and B1 cell specific protein","keratin 4","solute carrier family 23 member 1","neuronal PAS domain protein 2","LON peptidase N-terminal domain and ring finger 2","nudix hydrolase 9","hydroxysteroid 17-beta dehydrogenase 13","proliferation-associated 2G4","ELOVL fatty acid elongase 6","6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3","transmembrane channel like 7","ADP ribosylation factor like GTPase 6 interacting protein 1","serpin family B member 9","small cell adhesion glycoprotein","embigin","SIX homeobox 2","DLG associated protein 1","signal transducer and activator of transcription 2","NudC domain containing 2","interferon regulatory factor 2 binding protein 1","heat shock protein family A (Hsp70) member 4","forkhead box A3","solute carrier family 26 member 5","COMM domain containing 5","gametocyte specific factor 1","zinc finger protein 16","armadillo repeat containing 10","ring finger protein 34","acylphosphatase 2","TraB domain containing","activating transcription factor 7","RAS p21 protein activator 4B","suppressor of cytokine signaling 6","zinc finger protein 296","tubulin tyrosine ligase like 6","DNA polymerase eta","potassium voltage-gated channel modifier subfamily S member 3","kinesin family member 5B","A-kinase anchoring protein 13","cell division cycle associated 4","short chain dehydrogenase/reductase family 16C member 5","DPY30 domain containing 1","olfactory receptor family 10 subfamily A member 2","coiled-coil-helix-coiled-coil-helix domain containing 7","forkhead box N2","leiomodin 2","ubiquitin specific peptidase 32","carboxyl ester lipase","protein phosphatase, Mg2+/Mn2+ dependent 1D","G protein-coupled receptor 27","Morf4 family associated protein 1 like 1 pseudogene","kelch repeat and BTB domain containing 2","ribosomal oxygenase 2","TP53 regulated inhibitor of apoptosis 1","LSM3 homolog, U6 small nuclear RNA and mRNA degradation associated","KIAA0232","MTSS I-BAR domain containing 1","transmembrane protein 43","ring finger protein 139","ribosomal protein S9","tRNA splicing endonuclease subunit 34","glutathione S-transferase alpha 4","Myb/SANT DNA binding domain containing 4 with coiled-coils","NADH:ubiquinone oxidoreductase subunit A3","osteoclast associated Ig-like receptor","progestin and adipoQ receptor family member 8","nudix hydrolase 6","tetratricopeptide repeat, ankyrin repeat and coiled-coil containing 2","olfactory receptor family 7 subfamily G member 2","PKHD1 ciliary IPT domain containing fibrocystin/polyductin","olfactory receptor family 1 subfamily M member 1","DnaJ heat shock protein family (Hsp40) member C24","methyl-CpG binding domain protein 3 like 1","zinc finger protein 160","zinc finger protein 415","caveolae associated protein 3","CEA cell adhesion molecule 3","doublecortin domain containing 1","hyaluronan synthase 2","platelet derived growth factor D","sphingosine-1-phosphate receptor 1","heparan sulfate 6-O-sulfotransferase 2","pygopus family PHD finger 1","leucine rich repeat containing 8 VRAC subunit E","cAMP-dependent protein kinase inhibitor alpha","XK related 6","t-SNARE domain containing 1","formyl peptide receptor 2","formyl peptide receptor 1","fasciculation and elongation protein zeta 2","SRY-box transcription factor 7","chromosome 8 open reading frame 74","chromosome 11 open reading frame 24","ALK receptor tyrosine kinase","kynurenine aminotransferase 1","myotubularin 1","tRNA methyltransferase 61B","insulin receptor","mitofusin 1","GTPase, IMAP family member 8","neurturin","potassium calcium-activated channel subfamily M regulatory beta subunit 3","fucosyltransferase 3 (Lewis blood group)","potassium voltage-gated channel modifier subfamily G member 3","ATPase H+ transporting V0 subunit e2","protein kinase C epsilon","jagunal homolog 1","transcriptional adaptor 3","suppressor of cytokine signaling 5","C1GALT1 specific chaperone 1","chromosome 9 open reading frame 16","MORN repeat containing 4","zinc finger protein 672","zinc finger protein 692","nuclear apoptosis inducing factor 1","ribokinase","olfactory receptor family 2 subfamily M member 4","glutamate ionotropic receptor kainate type subunit 1","mucin 7, secreted","transmembrane protein 126A","transmembrane protein 126B","tripartite motif containing 8","neuropilin and tolloid like 2","claudin 20","CDC42 binding protein kinase gamma","SCAN domain containing 1","JunB proto-oncogene, AP-1 transcription factor subunit","family with sequence similarity 241 member B","transmembrane protein 37","UDP glucuronosyltransferase family 2 member B7","leucine rich alpha-2-glycoprotein 1","SHC binding and spindle associated 1","sclerostin domain containing 1","neuronal pentraxin 1","family with sequence similarity 98 member B","zinc finger protein 439","zinc finger protein 440","glucosidase alpha, acid","calcium activated nucleotidase 1","potassium two pore domain channel subfamily K member 3","zinc finger DHHC-type containing 16","carbohydrate sulfotransferase 11","exosome component 1","phosphoglycerate mutase 1","chromodomain helicase DNA binding protein 7","establishment of sister chromatid cohesion N-acetyltransferase 2","keratin 19","keratin 15","leucine rich adaptor protein 1","chloride voltage-gated channel 5","tubulin polymerization promoting protein","potassium voltage-gated channel subfamily D member 3","keratin 13","phosphodiesterase 7B","mitochondrial ribosomal protein L36","zinc finger protein 581","N-acetyltransferase 1","keratin 20","kinase suppressor of ras 2","zinc finger protein 524","MCC regulator of WNT signaling pathway","zinc finger and BTB domain containing 26","dermatan sulfate epimerase like","RNA polymerase I and III subunit C","ASXL transcriptional regulator 1","delta like non-canonical Notch ligand 2","zinc finger protein 562","zinc finger protein 318","zinc finger protein 561","WAS/WASL interacting protein family member 2","HOP homeobox","leucine rich repeat containing 8 VRAC subunit C","ribosomal L1 domain containing 1","leucine rich repeat containing 8 VRAC subunit D","peptidylprolyl isomerase D","collagen type XXIV alpha 1 chain","electron transfer flavoprotein dehydrogenase","lysophosphatidic acid receptor 3","prostaglandin E receptor 4","tubulin folding cofactor A","neuronal differentiation 2","microtubule associated protein 6","BCL2 like 1","olfactory receptor family 2 subfamily AT member 4","pleiotropic regulator 1","zinc finger protein 584","dynein axonemal intermediate chain 2","neuromedin U receptor 1","calsyntenin 1","CXXC finger protein 5","zinc finger protein 274","phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta","solute carrier family 25 member 33","ectodermal-neural cortex 1","splA/ryanodine receptor domain and SOCS box containing 1","pyrimidinergic receptor P2Y6","bromodomain PHD finger transcription factor","zinc finger protein interacting with K protein 1","G protein-coupled receptor 82","G protein-coupled receptor 34","pleckstrin homology and RhoGEF domain containing G5","activating transcription factor 7 interacting protein","LKAAEAR motif containing 1","regulator of G protein signaling 19","transcription elongation factor A2","defensin beta 4A","histone deacetylase 3","spermatogenesis associated 46","gephyrin","transmembrane protein 51","calmodulin binding transcription activator 1","galectin 4","leucine rich repeat containing 34","spermatogenesis associated 5 like 1","glycine amidinotransferase","RAS guanyl releasing protein 4","nescient helix-loop-helix 1","schlafen like 1","BCL2 apoptosis regulator","RAD9-HUS1-RAD1 interacting nuclear orphan 1","CTP synthase 1","kinase non-catalytic C-lobe domain containing 1","methyltransferase like 18","cilia and flagella associated protein 46","collagen type VIII alpha 2 chain","PWWP domain containing 2B","protocadherin beta 1","zinc finger protein 540","angiopoietin like 7","F-box and leucine rich repeat protein 14","exosome component 10","zinc finger protein 570","ninjurin 2","MLLT3 super elongation complex subunit","family with sequence similarity 90 member A1","ribonucleotide reductase regulatory subunit M2","trafficking protein particle complex 12","ribosomal protein S21","complement C3a receptor 1","mitochondrial rRNA methyltransferase 3","phosphatase and tensin homolog","ribosomal protein S7","prion like protein doppel","ribonuclease H1","prion protein","Kruppel like factor 17","FERM domain containing 5","cytochrome P450 family 4 subfamily F member 11","talin 2","galectin 9C","trans-golgi network vesicle protein 23 homolog B","F-box and WD repeat domain containing 10","zinc finger protein 217","SLIT-ROBO Rho GTPase activating protein 2C","ATP synthase mitochondrial F1 complex assembly factor 2","forkhead box B1","peptidylprolyl isomerase H","dynein regulatory complex subunit 3","zinc finger protein 57","shieldin complex subunit 1","jumonji domain containing 1C","lactate dehydrogenase A like 6B","synaptopodin","zinc finger protein 556","mal, T cell differentiation protein","zinc finger protein 554","RAB33B, member RAS oncogene family","thimet oligopeptidase 1","ankyrin repeat domain 20 family member A4","laminin subunit beta 2","ubiquitin specific peptidase 19","glutaminyl-tRNA synthetase 1","ORMDL sphingolipid biosynthesis regulator 3","small EDRK-rich factor 1A","Kruppel like factor 11","survival of motor neuron 1, telomeric","eukaryotic translation initiation factor 2 alpha kinase 3","MOB kinase activator 3A","lysine rich coiled-coil 1","NME/NM23 nucleoside diphosphate kinase 6","cytochrome c, somatic","CD8b molecule","schlafen family member 12","solute carrier family 9 member C1","olfactory receptor family 1 subfamily A member 1 (gene/pseudogene)","FERM domain containing 3","syntrophin beta 1","MDM2 binding protein","transcription elongation factor, mitochondrial","mitochondrial ribosomal protein L13","MALT1 paracaspase","interferon stimulated exonuclease gene 20","membrane bound O-acyltransferase domain containing 1","inhibitor of DNA binding 4, HLH protein","G protein-coupled receptor 22","C-X-C motif chemokine receptor 6","CCAAT enhancer binding protein beta","azurocidin 1","tryptase alpha/beta 1","poly(A) binding protein interacting protein 1","C-type lectin domain containing 7A","chromosome 5 open reading frame 34","neuronal growth regulator 1","zinc finger protein 131","mono-ADP ribosylhydrolase 2","dolichyl-phosphate N-acetylglucosaminephosphotransferase 1","basigin (Ok blood group)","histone H4 transcription factor","ceramide synthase 6","serine palmitoyltransferase long chain base subunit 3","coordinator of PRMT5 and differentiation stimulator","TP53 regulating kinase","beta-1,3-galactosyltransferase 1","olfactory receptor family 5 subfamily A member 1","C-type lectin domain family 12 member A","olfactory receptor family 5 subfamily A member 2","bisphosphoglycerate mutase","POP7 homolog, ribonuclease P/MRP subunit","ALG14 UDP-N-acetylglucosaminyltransferase subunit","succinate-CoA ligase GDP-forming beta subunit","StAR related lipid transfer domain containing 5","cold shock domain containing C2","regulator of calcineurin 2","interleukin 16","G protein subunit beta 2","cilia and flagella associated protein 53","olfactory receptor family 5 subfamily B member 2","MAPK regulated corepressor interacting protein 2","PDZ domain containing 3","C2CD2 like","aryl hydrocarbon receptor nuclear translocator 2","G protein subunit gamma 12","serine protease 27","synaptopodin 2","DnaJ heat shock protein family (Hsp40) member B7","cleavage and polyadenylation factor I subunit 1","EF-hand calcium binding domain 3","tetratricopeptide repeat domain 36","radial spoke head component 9","COP9 signalosome subunit 9","GTP binding protein 2","FGGY carbohydrate kinase domain containing","interleukin 17D","fucosyltransferase 9","transcription elongation factor A like 1","zinc finger protein 24","heat shock transcription factor Y-linked 1","mannosidase endo-alpha","RAB40A, member RAS oncogene family","mab-21 like 4","alanine--glyoxylate and serine--pyruvate aminotransferase","AF4/FMR2 family member 1","FGF1 intracellular binding protein","carnosine synthase 1","olfactory receptor family 10 subfamily H member 5","BTG3 associated nuclear protein","protein phosphatase 1 catalytic subunit alpha","host cell factor C1","cathepsin W","NIPA like domain containing 4","mucin like 1","syntrophin gamma 2","phosphodiesterase 3A","RAS guanyl releasing protein 1","kelch like family member 6","coiled-coil-helix-coiled-coil-helix domain containing 1","mitochondrial ribosomal protein L52","sphingomyelin phosphodiesterase acid like 3A","Rho family GTPase 1","RAD9 checkpoint clamp component A","EGF containing fibulin extracellular matrix protein 2","ArfGAP with GTPase domain, ankyrin repeat and PH domain 5","WASH complex subunit 2C","transmembrane protein 134","zinc finger matrin-type 3","zinc finger AN1-type containing 4","thymocyte selection associated","zinc finger protein 738","membrane spanning 4-domains A10","schlafen family member 11","family with sequence similarity 71 member D","coronin 1B","fucosyltransferase 10","leucine rich repeat containing 20","MUS81 structure-specific endonuclease subunit","transmembrane protein 217","olfactory receptor family 4 subfamily D member 9","zinc finger protein 596","cofilin 1","transmembrane and coiled-coil domain family 1","N(alpha)-acetyltransferase 16, NatA auxiliary subunit","EF-hand calcium binding domain 12","proteasome activator subunit 3 interacting protein 1","RAB43, member RAS oncogene family","fatty acid desaturase 6","COBW domain containing 1","RAB37, member RAS oncogene family","decapping mRNA 2","sorting nexin 32","ribosomal protein L38","cytochrome P450 family 7 subfamily B member 1","ovo like transcriptional repressor 1","retinoic acid receptor gamma","carboxylesterase 4A","carboxylesterase 3","slingshot protein phosphatase 3","carboxylesterase 2","pyruvate dehyrogenase phosphatase catalytic subunit 2","Sp3 transcription factor","Dmx like 1","methionyl aminopeptidase type 1D, mitochondrial","zinc finger protein 621","EGF like domain multiple 7","NAD synthetase 1","7-dehydrocholesterol reductase","neurobeachin","ribonuclease H2 subunit C","myeloma overexpressed","ankyrin repeat domain 13D","MYD88 innate immune signal transduction adaptor","oxidative stress responsive kinase 1","solute carrier family 22 member 13","PHD finger protein 8","lysocardiolipin acyltransferase 1","alcohol dehydrogenase 6 (class V)","lysine acetyltransferase 5","SH3 domain containing ring finger 3","glucoside xylosyltransferase 2","dephospho-CoA kinase domain containing","transcriptional adaptor 2B","coiled-coil domain containing 96","G protein-coupled receptor kinase 2","RELA proto-oncogene, NF-kB subunit","EvC ciliary complex subunit 2","zinc finger protein 680","HECT domain E3 ubiquitin protein ligase 4","family with sequence similarity 222 member B","basonuclin 2","heparanase","coenzyme Q2, polyprenyltransferase","heat shock protein family A (Hsp70) member 6","tRNA methyltransferase subunit 11-2","leucine rich repeat neuronal 3","lysine demethylase 2A","acyl-CoA synthetase medium chain family member 6","aarF domain containing kinase 5","mitochondrial ribosomal protein L57","NOC3 like DNA replication regulator","estrogen related receptor alpha","ras homolog family member D","copper metabolism domain containing 1","Ras association (RalGDS/AF-6) and pleckstrin homology domains 1","metaxin 1","adenylate cyclase 5","poly(ADP-ribose) polymerase family member 14","cysteinyl leukotriene receptor 1","poly(ADP-ribose) polymerase family member 15","CDC28 protein kinase regulatory subunit 1B","ATP binding cassette subfamily D member 2","actin binding LIM protein family member 3","mab-21 like 3","major facilitator superfamily domain containing 4B","VANGL planar cell polarity protein 1","glutaredoxin","IQ motif containing B1","synaptotagmin 12","golgin B1","doublesex and mab-3 related transcription factor 2","zinc finger protein 483","solute carrier family 2 member 14","G protein-coupled receptor 137","synuclein gamma","multimerin 2","mitotic spindle organizing protein 2A","tankyrase","zinc finger protein 449","zinc finger and BTB domain containing 21","protein phosphatase 1 regulatory subunit 3B","olfactory receptor family 10 subfamily K member 1","storkhead box 2","mitogen-activated protein kinase kinase kinase 11","tribbles pseudokinase 1","potassium two pore domain channel subfamily K member 7",null,"complement C1q B chain","complement C1q A chain","neuron derived neurotrophic factor","IQ motif containing F1","oxidized low density lipoprotein receptor 1","GLIPR1 like 1","dystroglycan 1","DAB adaptor protein 1","ARV1 homolog, fatty acid homeostasis modulator","N(alpha)-acetyltransferase 20, NatB catalytic subunit","coiled-coil domain containing 36","serum amyloid A1","mitochondrial contact site and cristae organizing system subunit 10","EH domain binding protein 1 like 1","THAP domain containing 2","ring finger protein 26","protein phosphatase 1 regulatory inhibitor subunit 14B","zinc ribbon domain containing 2","anterior gradient 3, protein disulphide isomerase family member","SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily c member 1","zinc finger protein 417","protein tyrosine phosphatase receptor type M","FKBP prolyl isomerase 2","vascular endothelial growth factor B","pseudopodium enriched atypical kinase 1","TNF receptor superfamily member 10d","macrophage stimulating 1","TNF receptor superfamily member 10c","GDP-mannose pyrophosphorylase B","MOB kinase activator 1B","zinc finger protein 622","chondroitin sulfate proteoglycan 4","sorting nexin 33","family with sequence similarity 166 member C","nucleic acid binding protein 1","adhesion G protein-coupled receptor F3","chromodomain helicase DNA binding protein 2","X-C motif chemokine receptor 1","coiled-coil domain containing 106","C-C motif chemokine receptor 9","centrosomal protein 83","sulfotransferase family 1B member 1","nudix hydrolase 4","pyruvate carboxylase","UDP glucuronosyltransferase family 2 member A1 complex locus","suppressor of cancer cell invasion","nicotinamide nucleotide adenylyltransferase 1","leucine rich repeat and fibronectin type III domain containing 4","trafficking protein particle complex 3 like","apolipoprotein B mRNA editing enzyme catalytic polypeptide like 4","solute carrier family 19 member 1","heat shock protein family B (small) member 7","Ras converting CAAX endopeptidase 1","ubiquinol-cytochrome c reductase hinge protein","taste 1 receptor member 1","eukaryotic translation initiation factor 1A X-linked","speedy/RINGO cell cycle regulator family member E2B","BCLAF1 and THRAP3 family member 3","proteasome 26S subunit, non-ATPase 1","adhesion G protein-coupled receptor G2","mucin 13, cell surface associated","sushi domain containing 5","heart development protein with EGF like domains 1","chromosome 11 open reading frame 80","translocase of outer mitochondrial membrane 20","chromosome 1 open reading frame 100","ArfGAP with FG repeats 1","signal transducer and activator of transcription 5B","CD7 molecule","2',3'-cyclic nucleotide 3' phosphodiesterase","junction plakoglobin","huntingtin associated protein 1","tudor domain containing 12","eukaryotic translation initiation factor 1","endonuclease V","ring finger protein 213","tigger transposable element derived 3","potassium voltage-gated channel subfamily H member 6","membrane associated ring-CH-type finger 10","polo like kinase 3","neuroepithelial cell transforming 1","dpy-19 like C-mannosyltransferase 1","DET1 partner of COP1 E3 ubiquitin ligase","phosphoethanolamine/phosphocholine phosphatase 1","zinc finger protein 791","polyhomeotic homolog 3","G protein-coupled receptor 160","spectrin beta, non-erythrocytic 2","golgi integral membrane protein 4","RNA binding motif protein 4B","ATP synthase membrane subunit DAPIT","homeobox B2","C1q and TNF related 1","membrane associated ring-CH-type finger 3","SWIM-type zinc finger 7 associated protein 1","solute carrier organic anion transporter family member 4C1","RNA binding motif protein 4","primary cilia formation","xyloside xylosyltransferase 1","UBX domain protein 2A","retina and anterior neural fold homeobox 2","leucine rich repeat containing 63","copper chaperone for superoxide dismutase","negative regulator of reactive oxygen species","centrosomal protein 19","kelch like family member 15","F-box protein 45","G protein subunit gamma 5","solute carrier family 25 member 30","chromosome 9 open reading frame 131","CD34 molecule","cathepsin F","methionine sulfoxide reductase B3","LEM domain containing 3","chromosome 16 open reading frame 91","toll like receptor 10","toll like receptor 1","toll like receptor 6","family with sequence similarity 174 member A","repulsive guidance molecule BMP co-receptor b","family with sequence similarity 53 member A","cytochrome b561 family member D1","glutathione S-transferase alpha 3","zinc finger DHHC-type containing 24","tRNA methyltransferase 10C, mitochondrial RNase P subunit","selectin P","cytosolic thiouridylase subunit 2","MAX dimerization protein MGA","chromosome 12 open reading frame 66","ADP ribosylation factor like GTPase 13A","sorting nexin 31","phosphatidylinositol glycan anchor biosynthesis class G","pre-mRNA processing factor 8","adenylate cyclase 6","phosphatidylinositol transfer protein alpha","DEAD-box helicase 23","zinc finger protein 80","zinc finger HIT-type containing 2","zinc finger and BTB domain containing 4","tyrosine kinase non receptor 1","zinc fingers and homeoboxes 3","pleckstrin homology like domain family A member 3","disrupted in renal carcinoma 1","solute carrier family 16 member 11","solute carrier family 16 member 13","cholinergic receptor nicotinic alpha 9 subunit","chromosome 11 open reading frame 45","exonuclease 1","Ral GTPase activating protein catalytic alpha subunit 1","DNA ligase 4","GTF2I repeat domain containing 2B","actin binding Rho activating protein","ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2","zwilch kinetochore protein","ribosomal protein L4","small nuclear RNA activating complex polypeptide 5","chromosome 12 open reading frame 76","contactin associated protein like 2","Bardet-Biedl syndrome 1","DENN domain containing 4A","germinal center associated signaling and motility","ankyrin repeat domain 36C","solute carrier family 26 member 9","major facilitator superfamily domain containing 4A","pellino E3 ubiquitin protein ligase family member 3","myosin IH","mitochondrial ribosomal protein L11","interleukin 20 receptor subunit beta","golgi transport 1A","akirin 1","MSL complex subunit 2","zinc finger protein 497","chemerin chemokine-like receptor 1","angel homolog 2","UDP glycosyltransferase 8","kyphoscoliosis peptidase","IQ motif containing K","solute carrier organic anion transporter family member 2A1","zinc finger protein 266","olfactory receptor family 7 subfamily D member 4","solute carrier family 29 member 2","BR serine/threonine kinase 2","beta-1,4-glucuronyltransferase 1","transmembrane protein 167A","SH3 and PX domains 2B","retroelement silencing factor 1","La ribonucleoprotein 7, transcriptional regulator","fibroblast growth factor binding protein 3","nuclear receptor subfamily 1 group D member 2","BRMS1 transcriptional repressor and anoikis regulator","ribosomal protein L15","family with sequence similarity 241 member A","HRas proto-oncogene, GTPase","WD repeat domain 49","signal recognition particle 72","Ras and Rab interactor 1","odontogenesis associated phosphoprotein","THAP domain containing 6","centrosomal protein 135","frizzled class receptor 4","betacellulin","PDZ domain containing 1","adhesion G protein-coupled receptor E1","DENN domain containing 6A","phosphodiesterase 12","glomulin, FKBP associated protein","dynein axonemal heavy chain 12","Yip1 interacting factor homolog A, membrane trafficking protein","cornichon family AMPA receptor auxiliary protein 2","amylase alpha 1B","NLR family pyrin domain containing 6","NADH:ubiquinone oxidoreductase subunit A11","arginine and serine rich coiled-coil 1","cation channel sperm associated auxiliary subunit delta","RAB1B, member RAS oncogene family","phosphatidylserine synthase 2","mitochondrial contact site and cristae organizing system subunit 13","chromosome 3 open reading frame 33","seizure related 6 homolog like 2","potassium channel tetramerization domain containing 13","purinergic receptor P2Y14","archaelysin family metallopeptidase 1","G protein-coupled receptor 171","CD164 molecule like 2","fucosyltransferase 1 (H blood group)","DEAH-box helicase 36","F-box and WD repeat domain containing 8","zymogen granule protein 16","kinesin light chain 2","solute carrier family 22 member 1","C-terminal binding protein 2","carbohydrate sulfotransferase 2","zinc finger DHHC-type containing 14","ATR serine/threonine kinase","ubiquitin conjugating enzyme E2 C","glycerol kinase 5","valosin containing protein interacting protein 1","PDLIM1 interacting kinase 1 like","TNF receptor associated factor 6","zinc finger protein 654","trans-golgi network vesicle protein 23 homolog C","mitochondrial ribosomal protein S22","phosphofurin acidic cluster sorting protein 1","MARCKS like 1","SH3 binding domain protein 5 like","yippee like 2","cell adhesion molecule 2","ABO, alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase","proteasome 26S subunit, non-ATPase 2","protein phosphatase, Mg2+/Mn2+ dependent 1E","family with sequence similarity 131 member A","cysteine and glycine rich protein 2","presenilin associated rhomboid like","DNA damage inducible transcript 3","propionyl-CoA carboxylase subunit alpha","dynactin subunit 2","zinc finger protein 408","CTD small phosphatase 2","cytoskeleton associated protein 5","Rho GTPase activating protein 1","mediator complex subunit 16","autophagy related 13","chromosome 1 open reading frame 127","carbohydrate sulfotransferase 1","golgin A8 family member A","von Willebrand factor A domain containing 3A","tumor protein p53 inducible protein 11","centromere protein S","dolichol kinase","phytanoyl-CoA dioxygenase domain containing 1","cyclin E2","5-phosphohydroxy-L-lysine phospho-lyase","ankyrin repeat and sterile alpha motif domain containing 4B","cystatin E/M","GRAM domain containing 2A","zinc finger protein 519","LSM1 homolog, mRNA degradation associated","barrier to autointegration factor 1","apolipoprotein F","cholinergic receptor nicotinic alpha 7 subunit","TMEM9 domain family member B","nuclear receptor interacting protein 3","protein tyrosine phosphatase non-receptor type 2","signal peptide, CUB domain and EGF like domain containing 2","eukaryotic translation initiation factor 1A domain containing","SMAD family member 2","eukaryotic translation initiation factor 3 subunit F","zinc finger protein 25","ADP ribosylation factor like GTPase 10","clathrin light chain B","proprotein convertase subtilisin/kexin type 1","Rieske Fe-S domain containing","coiled-coil domain containing 14","TBC1 domain family member 10C","spliceosome associated factor 1, recruiter of U4/U6.U5 tri-snRNP","protein phosphatase 2 regulatory subunit Bdelta","multiple C2 and transmembrane domain containing 1","DNA polymerase delta 4, accessory subunit","leucine rich repeat containing 25","dipeptidyl peptidase like 10","cardiotrophin like cytokine factor 1","ubiquilin like","ubiquilin 3","lipoyl(octanoyl) transferase 2","potassium voltage-gated channel subfamily E regulatory subunit 3","calcium binding protein 4","ALG10 alpha-1,2-glucosyltransferase B","DR1 associated protein 1","LON peptidase N-terminal domain and ring finger 3","uncoupling protein 3","uncoupling protein 2","chromosome 11 open reading frame 68","proteasomal ATPase associated factor 1","mitochondrial ribosomal protein L48","RAB6A, member RAS oncogene family","purinergic receptor P2Y2","FOS like 1, AP-1 transcription factor subunit","ERCC excision repair 4, endonuclease catalytic subunit","coiled-coil domain containing 85B","transmembrane protein 70","ribosomal protein S6 kinase B2","RecQ mediated genome instability 2","target of myb1 like 2 membrane trafficking protein","testis expressed 26","zinc finger protein 77","G protein-coupled receptor 156","mitoregulin","keratinocyte differentiation factor 1","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase like 1","MLX interacting protein","nuclear receptor subfamily 2 group F member 1","aurora kinase A interacting protein 1","tubulin tyrosine ligase like 11","translocase of outer mitochondrial membrane 5","solute carrier family 35 member E3","zinc finger protein 169","RuvB like AAA ATPase 1","stratifin","methionine sulfoxide reductase A","CTD nuclear envelope phosphatase 1","ETS variant transcription factor 4","SWI5 homologous recombination repair protein","GRB2 binding adaptor protein, transmembrane","BAR/IMD domain containing adaptor protein 2","cellular repressor of E1A stimulated genes 2","zinc finger DHHC-type containing 21","pleckstrin homology and FYVE domain containing 2","alpha-2-macroglobulin","ADP ribosylation factor like GTPase 4D","docking protein 7","leucine rich repeat neuronal 1","ubiquitin conjugating enzyme E2 O","ORAI calcium release-activated calcium modulator 3","unc-119 lipid binding chaperone B","DENN domain containing 2C","pleckstrin homology and coiled-coil domain containing D1","achaete-scute family bHLH transcription factor 3","tubulin beta 6 class V","LysM domain containing 3","adhesion molecule with Ig like domain 3","beta-1,3-galactosyltransferase 6","zinc finger protein 613","chromosome 11 open reading frame 16","transmembrane serine protease 7","nuclear protein 1, transcriptional regulator","janus kinase and microtubule interacting protein 2","metallo-beta-lactamase domain containing 2","taperin","zinc finger protein 683","solute carrier family 35 member A4","crystallin beta-gamma domain containing 2","inositol hexakisphosphate kinase 1","SS nuclear autoantigen 1","cleavage stimulation factor subunit 3","YES proto-oncogene 1, Src family tyrosine kinase","charged multivesicular body protein 6","transmembrane protein 39A","t-complex 11 like 1","glutathione peroxidase 2","coiled-coil domain containing 57","forkhead box G1","sphingosine kinase 1","BCL2 interacting protein 3","Myb related transcription factor, partner of profilin","leucine rich repeat transmembrane neuronal 4","ATPase family AAA domain containing 5","small integral membrane protein 19","zinc finger protein 404","rotatin","olfactory receptor family 4 subfamily K member 17","acyl-CoA binding domain containing 7","anaphase promoting complex subunit 2","olfactory receptor family 4 subfamily K member 13","zinc finger and BTB domain containing 8 opposite strand","solute carrier family 35 member G1","zinc finger protein 135","cytochrome c oxidase subunit 8A","tachykinin precursor 4","zinc finger and SCAN domain containing 2","proline rich 18","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 4","cell division cycle 26","hydroxysteroid 11-beta dehydrogenase 2","cytokine receptor like factor 3","arginyl aminopeptidase","EP300 interacting inhibitor of differentiation 2","DMRT like family A1","EP300 interacting inhibitor of differentiation 2B","regulating synaptic membrane exocytosis 2","potassium channel modulatory factor 1","DnaJ heat shock protein family (Hsp40) member C30","SPRY domain containing 4","spectrin repeat containing nuclear envelope family member 3","CDC like kinase 2","lysophosphatidylcholine acyltransferase 4","solute carrier organic anion transporter family member 3A1","zinc finger protein 575","WD repeat domain 25","SAGA complex associated factor 29","phospholipase A and acyltransferase 3","olfactory receptor family 5 subfamily AN member 1","pleckstrin homology like domain family B member 3","proline rich 15","G protein subunit gamma 7","upstream transcription factor family member 3","cyclin N-terminal domain containing 1","kelch repeat and BTB domain containing 11","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 5","mitogen-activated protein kinase kinase kinase 19","lamin B2","regulator of microtubule dynamics 1","mex-3 RNA binding family member C","ring finger protein 152","myosin ID","leucine rich repeat containing 37A","olfactory receptor family 4 subfamily F member 17","brain derived neurotrophic factor","coiled-coil domain containing 121","acyl-CoA synthetase family member 3","BCL2 family apoptosis regulator BOK","zinc finger protein 843","ribosomal biogenesis factor","profilin family member 4","cyclin dependent kinase 5 regulatory subunit 1","NCK associated protein 5","RUN and FYVE domain containing 1","brain abundant membrane attached signal protein 1","olfactory receptor family 51 subfamily L member 1","leucine rich repeat containing 37 member A3","V-set and immunoglobulin domain containing 10","iroquois homeobox 5","meteorin like, glial cell differentiation regulator","family with sequence similarity 91 member A1","WD repeat and SOCS box containing 2","glutamate ionotropic receptor NMDA type subunit 1","SRY-box transcription factor 11","thymidylate synthetase","peroxisomal membrane protein 2","transcription elongation factor A N-terminal and central domain containing","PNMA family member 1","transcriptional and immune response regulator","MEF2 activating motif and SAP domain containing transcriptional regulator","ankyrin repeat and LEM domain containing 2","fucosyltransferase 2","EF-hand calcium binding domain 5","glucosaminyl (N-acetyl) transferase 4","mucin 20, cell surface associated","THAP domain containing 4","nuclear factor of activated T cells 2 interacting protein","lymphocyte antigen 6 family member H","fin bud initiation factor homolog","family with sequence similarity 89 member B","serine hydroxymethyltransferase 1","dipeptidyl peptidase 7","SEC24 homolog C, COPII coat complex component","SMCR8-C9orf72 complex subunit","methylenetetrahydrofolate reductase","chromosome 19 open reading frame 18","DEAF1 transcription factor","metaxin 3","transmembrane protein 80","SIX homeobox 5","interferon omega 1","F-box protein 46","zinc finger DHHC-type containing 13","solute carrier family 38 member 9","alkaline ceramidase 2","WD repeat domain 73","DNA polymerase epsilon, catalytic subunit","PH domain containing endocytic trafficking adaptor 2","sodium voltage-gated channel beta subunit 4","DS cell adhesion molecule like 1","ras homolog family member G","EPS8 like 2","zinc finger DHHC-type containing 22","anoctamin 6","zinc finger and BTB domain containing 34","family with sequence similarity 9 member B","nudix hydrolase 4B","family with sequence similarity 210 member A","transaldolase 1","unc-51 like autophagy activating kinase 1","ribosomal modification protein rimK like family member A","clavesin 1","ribosomal protein S6 kinase A3","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 8","pseudouridine synthase 1","chromodomain helicase DNA binding protein 9","olfactory receptor family 2 subfamily T member 12","sperm acrosome associated 4","olfactory receptor family 2 subfamily T member 33","glutamine amidotransferase like class 1 domain containing 1","tripartite motif containing 72","mannosidase alpha class 1B member 1","potassium voltage-gated channel subfamily A member 3","frizzled class receptor 8","gap junction protein delta 4","F-box protein 39","potassium voltage-gated channel subfamily A member 2","DNA topoisomerase III alpha","CASK interacting protein 2","zinc finger and BTB domain containing 38","BEN domain containing 2","coiled-coil domain containing 71","LRRN4 C-terminal like","translocase of inner mitochondrial membrane 22","HIC ZBTB transcriptional repressor 1","PTPRF interacting protein alpha 3","MAGE family member F1","uromodulin like 1","sterile alpha motif domain containing 9 like","pro-apoptotic WT1 regulator","TGFB induced factor homeobox 1","mitochondrial elongation factor 2","nucleosome assembly protein 1 like 5","NIM1 serine/threonine protein kinase","CD19 molecule","glutamate rich 5","nuclear receptor subfamily 2 group C member 2","G protein-coupled receptor 4","acyl-CoA thioesterase 4","caveolae associated protein 1","ariadne RBR E3 ubiquitin protein ligase 2","RNA binding motif protein 44","zinc finger and BTB domain containing 33","zinc finger BED-type containing 2","iroquois homeobox 3","reprimo, TP53 dependent G2 arrest mediator homolog","olfactory receptor family 2 subfamily B member 11","solute carrier family 25 member 22","rabaptin, RAB GTPase binding effector protein 2","antioxidant 1 copper chaperone","transducin beta like 1 X-linked receptor 1","sterile alpha motif domain containing 12","CD163 molecule","chromosome 18 open reading frame 32","p53-induced death domain protein 1","zinc finger protein 491","ribosomal protein lateral stalk subunit P2","histone H3 associated protein kinase","Jun proto-oncogene, AP-1 transcription factor subunit","cleavage stimulation factor subunit 2 tau variant","piggyBac transposable element derived 5","chromosome 12 open reading frame 54","glucosylceramidase beta","acyl-CoA dehydrogenase family member 9","interleukin 17 receptor A","patatin like phospholipase domain containing 2","angiotensin II receptor associated protein","CD163 molecule like 1","serine/arginine repetitive matrix 3","THAP domain containing 5","calcium release activated channel regulator 2B","small ubiquitin like modifier 4","MAGE family member B10","DnaJ heat shock protein family (Hsp40) member C28","N-acetylated alpha-linked acidic dipeptidase like 2","CD151 molecule (Raph blood group)","RNA polymerase II subunit L","FAM20C golgi associated secretory pathway kinase","nectin cell adhesion molecule 3","solute carrier family 35 member G5","annexin A2 receptor","transmembrane protein 94","FLII actin remodeling protein","SRY-box transcription factor 12","heterogeneous nuclear ribonucleoprotein A0","potassium inwardly rectifying channel subfamily J member 10","chitinase domain containing 1","protocadherin beta 9","zinc finger protein 620","zinc finger protein 518A","transmembrane protein 187","small vasohibin binding protein","zinc finger protein 619","coiled-coil domain containing 184","adaptor related protein complex 3 subunit sigma 1","growth factor receptor bound protein 2","zinc finger and BTB domain containing 41","ubiquitin conjugating enzyme E2 N","ADP ribosylation factor like GTPase 6 interacting protein 6","zinc finger protein 354C","MAM domain containing 4","CENPB DNA-binding domain containing 1","outer dense fiber of sperm tails 3","Bet1 golgi vesicular membrane trafficking protein like","ribosomal protein S27","RIC8 guanine nucleotide exchange factor A","IMP U3 small nucleolar ribonucleoprotein 3","ankyrin repeat and SOCS box containing 8","lipocalin 15","outer dense fiber of sperm tails 3B","dpy-19 like 2","chromosome 2 open reading frame 73","TSPY like 6","leucine rich repeat containing 75B","DNA methyltransferase 1 associated protein 1","ADAMTS like 1","calcium homeostasis modulator family member 5","inosine monophosphate dehydrogenase 2","ALS2 C-terminal like","myeloid leukemia factor 1","NADH:ubiquinone oxidoreductase complex assembly factor 3","chromosome 2 open reading frame 69","GRAM domain containing 1C","signal transducing adaptor family member 2","testis specific serine kinase 6","bolA family member 1","phosphodiesterase 4D interacting protein","DEAD-box helicase 10","golgin A8 family member Q","protein phosphatase 1 regulatory subunit 42","NADH:ubiquinone oxidoreductase core subunit V2","DALR anticodon binding domain containing 3","zinc finger protein 114","zinc finger protein 518B","APC membrane recruitment protein 3","zinc finger protein 366","ligand dependent nuclear receptor corepressor like","par-6 family cell polarity regulator gamma","zinc finger protein 454","SH2B adaptor protein 1","zinc finger CCCH-type containing 12D","vomeronasal 1 receptor 1","protein O-glucosyltransferase 3","plectin","ring finger protein 212","serine protease 36","zinc finger protein 543","transmembrane protein 151B","polypeptide N-acetylgalactosaminyltransferase 11","WD repeat domain 6","GEN1 Holliday junction 5' flap endonuclease","transmembrane serine protease 9","aquaporin 11","transmembrane protein 11","zinc finger protein 354B","potassium voltage-gated channel modifier subfamily G member 2","calmodulin like 3","zinc finger AN1-type containing 2A","pleckstrin homology domain containing M3","zinc finger protein 223","family with sequence similarity 220 member A","DnaJ heat shock protein family (Hsp40) member C22","CEP295 N-terminal like","BEN domain containing 3","5'-nucleotidase domain containing 1","glycine decarboxylase","cytochrome c oxidase assembly factor COX14","minichromosome maintenance domain containing 2","prolyl 4-hydroxylase, transmembrane","urocortin 3","deltex E3 ubiquitin ligase 3","kelch like family member 11","cortexin 1","solute carrier family 25 member 20","carbonic anhydrase 8","CD28 molecule","EPM2A interacting protein 1","erb-b2 receptor tyrosine kinase 4","MAF bZIP transcription factor","catenin beta interacting protein 1","GTP binding protein 6 (putative)","endoplasmic reticulum to nucleus signaling 1","G protein-coupled receptor 35","chromosome 10 open reading frame 53","cysteine and serine rich nuclear protein 3","zinc finger protein 713","poly(ADP-ribose) polymerase family member 10","dynactin associated protein","SUZ12 polycomb repressive complex 2 subunit","NOP2/Sun RNA methyltransferase 3","potassium channel tetramerization domain containing 12","dihydrofolate reductase 2","ribonuclease P and MRP subunit p25","glutamate ionotropic receptor NMDA type subunit associated protein 1","thrombomodulin","glycoprotein V platelet","cytochrome c oxidase subunit 5A","syntaxin 19","family with sequence similarity 219 member B","zinc fingers and homeoboxes 2","carboxypeptidase N subunit 2","copine 7","CD300 molecule like family member b","glycerophosphodiester phosphodiesterase domain containing 4","regulatory subunit of type II PKA R-subunit domain containing 1","mannose phosphate isomerase","tripartite motif containing 73","5-oxoprolinase, ATP-hydrolysing","transmembrane protein 52","transmembrane protein 139","EF-hand calcium binding domain 13","musculin","apolipoprotein L domain containing 1","refilin A","exosome component 4","dpy-19 like C-mannosyltransferase 3","TATA-box binding protein associated factor 7","zinc finger protein 852","forkhead box E1","phosphoribosylformylglycinamidine synthase","hydroxypyruvate isomerase (putative)","cytochrome b-245 chaperone 1","tetrapeptide repeat homeobox 1","galectin 7","zinc finger protein 552","cyclin G associated kinase","zinc finger and BTB domain containing 7A","Tu translation elongation factor, mitochondrial","glutamate rich 3","RecQ mediated genome instability 1","CST telomere replication complex component 1","F-box protein 34","selenoprotein W","eukaryotic translation initiation factor 3 subunit K","Morf4 family associated protein 1 like 1","sorting nexin 18","aurora kinase B","chromosome 14 open reading frame 39","Morf4 family associated protein 1","chromosome 3 open reading frame 38","kelch domain containing 7A","transmembrane protein 107","ribosome biogenesis regulator 1 homolog","exocyst complex component 3 like 1","tripartite motif family like 2","regulator of chromosome condensation 2","immunoglobulin superfamily member 22","chromosome 9 open reading frame 50","ZFP42 zinc finger protein","coiled-coil domain containing 89","dolichyl-phosphate mannosyltransferase subunit 3, regulatory","cytochrome c1","period circadian regulator 1","transmembrane O-mannosyltransferase targeting cadherins 2","phenylalanyl-tRNA synthetase subunit alpha","SPT2 chromatin protein domain containing 1","chromosome 10 open reading frame 67","sterile alpha motif domain containing 4B","GTPase, IMAP family member 7","enhancer of mRNA decapping 3","T cell activation inhibitor, mitochondrial","alpha-L-fucosidase 1","gametogenetin","transmembrane protein 125","zinc finger protein 664","calreticulin","MAGE family member D1","GVQW motif containing 3","low density lipoprotein receptor class A domain containing 3","cadherin 4","RAD23 homolog A, nucleotide excision repair protein","photoreceptor cilium actin regulator","GADD45G interacting protein 1","DAN domain BMP antagonist family member 5","protein tyrosine phosphatase non-receptor type 11","NOP2/Sun RNA methyltransferase family member 7","family with sequence similarity 156 member B","RAB39A, member RAS oncogene family","CDC like kinase 3","major histocompatibility complex, class II, DQ beta 1","GATA binding protein 2","AT-rich interaction domain 3B","phosphofurin acidic cluster sorting protein 2","ELMO domain containing 2","early growth response 3","catsper channel auxiliary subunit epsilon","von Willebrand factor A domain containing 1","gem nuclear organelle associated protein 4","kelch like family member 28","makorin ring finger protein 3","zinc finger and BTB domain containing 18","chromosome 14 open reading frame 28","SHANK associated RH domain interactor","ladybird homeobox 2","dynein heavy chain domain 1","SLIT and NTRK like family member 4","GRIP and coiled-coil domain containing 1","leucine rich single-pass membrane protein 2","class II major histocompatibility complex transactivator","zinc finger protein, FOG family member 1","arachidonate 15-lipoxygenase type B","phospholipase D family member 6","CDC42 effector protein 4","olfactory receptor family 6 subfamily C member 4","zinc finger and BTB domain containing 42","laccase domain containing 1","MAF1 homolog, negative regulator of RNA polymerase III","Fc fragment of IgE receptor Ia","WD repeat domain 97","PC-esterase domain containing 1B","apolipoprotein B mRNA editing enzyme catalytic subunit 3B","pipecolic acid and sarcosine oxidase","family with sequence similarity 216 member B","myeloid associated differentiation marker","maestro heat like repeat family member 1","SERTA domain containing 2","A-kinase anchoring protein 5","NTPase KAP family P-loop domain containing 1","ring finger protein 227","Cbp/p300 interacting transactivator with Glu/Asp rich carboxy-terminal domain 4","ATP binding cassette subfamily A member 13","tigger transposable element derived 5","pyridoxal dependent decarboxylase domain containing 1","chromosome 1 open reading frame 194","zinc finger protein 154","R3H domain containing 2","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 3","neurexin 1","selenophosphate synthetase 2","G protein-coupled bile acid receptor 1","zinc finger protein 784","zinc finger protein 648","chromosome 14 open reading frame 119","golgin A8 family member J","Bardet-Biedl syndrome 10","FLT3 interacting zinc finger 1","poly(U) binding splicing factor 60","scavenger receptor cysteine rich family member with 5 domains","dCTP pyrophosphatase 1","zinc finger protein 771","teashirt zinc finger homeobox 1","phosphoseryl-tRNA kinase","suppressor of cytokine signaling 4","zinc binding alcohol dehydrogenase domain containing 2","zinc finger protein 48","ankyrin repeat domain 18A","WAP four-disulfide core domain 11","transmembrane protein 86B","septin 1","tRNA selenocysteine 1 associated protein 1","exocyst complex component 3","tudor domain containing 6","chromosome 12 open reading frame 40","casein kinase 1 alpha 1 like","Ly6/neurotoxin 1","mediator complex subunit 14","fumarylacetoacetate hydrolase domain containing 1","testis development related protein","regulator of chromosome condensation 1","protein activator of interferon induced protein kinase EIF2AK2","zinc and ring finger 2","retinal pigment epithelium-derived rhodopsin homolog","solute carrier family 9 member A4","zinc finger protein 816","FYVE, RhoGEF and PH domain containing 6","adhesion G protein-coupled receptor D2","phospholipase D family member 5","ornithine decarboxylase antizyme 2","patatin like phospholipase domain containing 1","coiled-coil domain containing 43","potassium channel tetramerization domain containing 4","meiosis specific with coiled-coil domain","frizzled class receptor 2","tigger transposable element derived 2","ITPR interacting domain containing 1","hematopoietic cell-specific Lyn substrate 1","maturin, neural progenitor differentiation regulator homolog","zinc finger protein 609","p21 (RAC1) activated kinase 2","coiled-coil domain containing 66","defensin beta 124","multiple coagulation factor deficiency 2","harbinger transposase derived 1","chromosome 11 open reading frame 71","cytochrome P450 family 8 subfamily B member 1","growth arrest specific 1","Rho GTPase activating protein 45","zinc finger protein 571","GLIPR1 like 2","mitoguardin 1","potassium voltage-gated channel subfamily E regulatory subunit 1","proline rich 26","nuclear receptor interacting protein 1","zinc finger and SCAN domain containing 4","ring finger protein 182","TSPY like 5","fucosyltransferase 7","H2A clustered histone 6","SKI/DACH domain containing 1","H2B clustered histone 4","Mab-21 domain containing 2","somatostatin receptor 2","zinc finger protein 594","polycomb group ring finger 5","solute carrier family 47 member 2","perforin 1","olfactory receptor family 2 subfamily A member 4","mab-21 like 1","YOD1 deubiquitinase","transmembrane protein 64","shisa family member 2","sphingosine-1-phosphate receptor 5","G protein-coupled receptor 157","solute carrier family 36 member 4","zinc finger DHHC-type containing 20","ankyrin repeat domain 30B","ZFP3 zinc finger protein","arylsulfatase family member J","mitogen-activated protein kinase kinase kinase 15","inorganic pyrophosphatase 1","proteasome assembly chaperone 4","MAP6 domain containing 1","zinc finger protein 443","C-X-C motif chemokine receptor 2","gremlin 2, DAN family BMP antagonist","signal sequence receptor subunit 4","calcyphosine 2","zinc finger protein 792","CUE domain containing 1","scribble planar cell polarity protein","potassium channel tetramerization domain containing 2","D-2-hydroxyglutarate dehydrogenase","oxytocin receptor","cap methyltransferase 2","family with sequence similarity 83 member H","olfactory receptor family 56 subfamily A member 1","zinc finger protein 572","suppressor of tumorigenicity 20","phosphatidylinositol transfer protein beta","transcription elongation factor A like 8","olfactory receptor family 52 subfamily E member 4","leucine rich repeat containing 57","mitochondrial ribosomal protein L14","G protein-coupled receptor 137C","Bardet-Biedl syndrome 12","ZFP82 zinc finger protein","olfactory receptor family 52 subfamily N member 5","leucine rich single-pass membrane protein 1","NAD(P)H quinone dehydrogenase 1","apoptosis enhancing nuclease","fukutin related protein","trafficking protein particle complex 5","rabphilin 3A like (without C2 domains)","solute carrier family 25 member 42","Fc receptor like 6","methyltransferase like 23","solute carrier family 26 member 11","HIG1 hypoxia inducible domain family member 1A","mitogen-activated protein kinase 15","euchromatic histone lysine methyltransferase 1","adiponectin, C1Q and collagen domain containing","coagulation factor II thrombin receptor","zinc finger protein 707","mucin 16, cell surface associated","nucleophosmin 1","praja ring finger ubiquitin ligase 1","dehydrogenase E1 and transketolase domain containing 1","chromosome 4 open reading frame 50","H2A.W histone","zinc finger protein 746","RNA polymerase II subunit A","TLC domain containing 5","FRAT regulator of WNT signaling pathway 2","transmembrane protein 102","zinc finger protein 322","NME/NM23 family member 9","hephaestin like 1","leucine rich repeat containing 75A","cilia and flagella associated protein 65","DExD/H-box 60 like","spectrin repeat containing nuclear envelope family member 4","2-oxoglutarate and iron dependent oxygenase domain containing 3","WASH complex subunit 1","apoptosis associated tyrosine kinase","dendrin","zinc finger protein 467","SRY-box transcription factor 2","zinc finger protein 678","transmembrane protein 45A","RAP2B, member of RAS oncogene family","zinc finger and BTB domain containing 2","ring finger protein 135","acyl-CoA binding domain containing 4","N-sulfoglucosamine sulfohydrolase","mab-21 like 2","FA complementation group B","SET domain containing 2, histone lysine methyltransferase","chromosome 6 open reading frame 223","mex-3 RNA binding family member D","mitochondrial ribosomal protein S23","follicular dendritic cell secreted protein","G protein-coupled receptor 135","SLX1 homolog B, structure-specific endonuclease subunit","ankyrin repeat domain 62","purinergic receptor P2Y13","TNF superfamily member 15","ZFP41 zinc finger protein","pleckstrin homology like domain family A member 2","autophagy related 9B","zinc finger protein 875","PLAG1 zinc finger","olfactory receptor family 8 subfamily H member 1","Yip1 domain family member 6","olfactory receptor family 5 subfamily T member 2","zinc finger and BTB domain containing 20","divergent protein kinase domain 2A","chromosome 5 open reading frame 30","adhesion molecule with Ig like domain 1","G protein-coupled receptor 3","olfactory receptor family 5 subfamily AS member 1","siah E3 ubiquitin protein ligase 2","coatomer protein complex subunit gamma 1","solute carrier family 9 member A9","LSM10, U7 small nuclear RNA associated","RELT like 1","regulatory factor X7","solute carrier family 35 member C1","T cell immunoreceptor with Ig and ITIM domains","ring finger protein 41","solute carrier family 2 member 4","iron-sulfur cluster assembly factor IBA57","claudin 7","zinc finger protein 329","zinc finger protein 101","chromosome 5 open reading frame 24","2-aminoethanethiol dioxygenase","cytochrome c oxidase assembly factor 4 homolog","olfactory receptor family 4 subfamily P member 4","protein kinase AMP-activated non-catalytic subunit gamma 1","GINS complex subunit 3","coiled-coil domain containing 149","mitochondrial ribosomal protein S11","small nuclear ribonucleoprotein polypeptide E","rhotekin 2","PNMA family member 8A","carbohydrate sulfotransferase 15","USH1 protein network component sans","MGAT4 family member C","isocitrate dehydrogenase (NADP(+)) 2","olfactory receptor family 52 subfamily A member 1","transmembrane protein 259","guided entry of tail-anchored proteins factor 1","trinucleotide repeat containing 18","family with sequence similarity 181 member B","transmembrane protein 30B","Dexi homolog","zinc finger protein 716","NOP10 ribonucleoprotein","tudor and KH domain containing","zinc finger protein 708","IST1 factor associated with ESCRT-III","ERCC excision repair 6 like 2","mitochondrial ribosomal protein L41","cAMP responsive element binding protein 3 like 2","P2Y receptor family member 8","unc-5 netrin receptor C","tRNA splicing endonuclease subunit 54","repulsive guidance molecule BMP co-receptor a","ankyrin repeat and SOCS box containing 18","ubiquitin like modifier activating enzyme 7","mitochondrial ribosomal protein S16","shisa like 2A","RAD51 paralog B","LDOC1 regulator of NFKB signaling","ADP ribosylation factor like GTPase 6 interacting protein 4","exostosin glycosyltransferase 1","serine hydroxymethyltransferase 2","MOB kinase activator 2","HHIP like 1","ATPase H+ transporting accessory protein 2","cytochrome b5 domain containing 1","family with sequence similarity 153 member B","beta-secretase 2","ubiquitin conjugating enzyme E2 E2","synemin","gamma-aminobutyric acid type A receptor gamma3 subunit","NLR family pyrin domain containing 10","izumo sperm-egg fusion 1","adaptor related protein complex 1 subunit sigma 2","chromosome 8 open reading frame 33","DDB1 and CUL4 associated factor 4 like 1","sperm acrosome associated 6","zinc finger and SCAN domain containing 22","potassium inwardly rectifying channel subfamily J member 14","F-box and leucine rich repeat protein 6","complement C1s","glycolipid transfer protein domain containing 2","KIAA2012","kelch repeat and BTB domain containing 3","ybeY metalloendoribonuclease","CLN8 transmembrane ER and ERGIC protein","phosphatidylinositol specific phospholipase C X domain containing 1","calcium voltage-gated channel auxiliary subunit beta 4","trafficking protein particle complex 6B","piggyBac transposable element derived 4","NPL4 homolog, ubiquitin recognition factor","teashirt zinc finger homeobox 2","calpain 12","exocyst complex component 7","karyopherin subunit alpha 2","XK related X-linked","centrosomal protein 97","FES proto-oncogene, tyrosine kinase","glutaredoxin 5","family with sequence similarity 104 member B","matrix remodeling associated 7","LIM domain kinase 2","major facilitator superfamily domain containing 5","ribonuclease A family member 10 (inactive)","acireductone dioxygenase 1","RWD domain containing 4","sphingolipid transporter 3 (putative)","SATB homeobox 1","neurexophilin 3","colony stimulating factor 1 receptor","EPH receptor B3","actin like 10","epithelial mitogen","secondary ossification center associated regulator of chondrocyte maturation","trafficking kinesin protein 1","tetraspanin 10","olfactory receptor family 2 subfamily V member 2","phospholipase C beta 1","spindle and kinetochore associated complex subunit 2","necdin, MAGE family member","tetratricopeptide repeat domain 3","BRICHOS domain containing 5","IgA inducing protein","tsukushi, small leucine rich proteoglycan","C-X9-C motif containing 4","annexin A2","progestin and adipoQ receptor family member 7","pappalysin 1","MAF bZIP transcription factor A","neugrin, neurite outgrowth associated","ribosomal protein S17","hydroxycarboxylic acid receptor 2","coiled-coil domain containing 87","chromosome 1 open reading frame 116","cysteine rich protein 2","DEAD-box helicase 28","acyl-CoA binding domain containing 3","chromosome 16 open reading frame 72","phosphatidylinositol specific phospholipase C X domain containing 3","vitelline membrane outer layer 1 homolog","olfactory receptor family 4 subfamily F member 15","ALG12 alpha-1,6-mannosyltransferase","LCK proto-oncogene, Src family tyrosine kinase","collagen type XVIII alpha 1 chain","RNA binding motif protein 10","adhesion G protein-coupled receptor G3","ribosomal protein L35a","solute carrier family 25 member 18","zinc finger protein 721","chromosome 11 open reading frame 54","centrosomal protein 63","SRP receptor subunit alpha","EWS RNA binding protein 1","high mobility group nucleosomal binding domain 4","spermatogenesis associated 13","gap junction protein gamma 1","CCR4-NOT transcription complex subunit 10","metastasis associated 1","zinc finger protein 662","cell adhesion molecule 1","zinc finger protein 320","chromosome 12 open reading frame 60","pyrroline-5-carboxylate reductase 1","N(alpha)-acetyltransferase 38, NatC auxiliary subunit","sphingolipid transporter 2","mast cell expressed membrane protein 1","adaptor related protein complex 2 subunit alpha 2","solute carrier family 8 member A1","solute carrier family 25 member 21","4-aminobutyrate aminotransferase","solute carrier family 25 member 10","calcium/calmodulin dependent protein kinase ID","RANBP2 like and GRIP domain containing 6","LysM domain containing 4","WBP2 N-terminal like","immunoglobulin superfamily member 5","arylformamidase","growth arrest specific 6","nebulin","brain enriched guanylate kinase associated","Rho guanine nucleotide exchange factor 37","CUB and Sushi multiple domains 1","centrosomal protein 57 like 1","ripply transcriptional repressor 3","ankyrin repeat domain 20 family member A2","gap junction protein delta 3","RAB interacting factor","transmembrane protein 119","FA complementation group F","single-pass membrane protein with aspartate rich tail 1","gamma-aminobutyric acid type A receptor rho3 subunit (gene/pseudogene)","carbohydrate sulfotransferase 6","POTE ankyrin domain family member C","RuvB like AAA ATPase 2","GDP-D-glucose phosphorylase 1","RIMS binding protein 3C","proline rich 36","PTTG1 interacting protein","DEAD-box helicase 41","abhydrolase domain containing 16B","coiled-coil domain containing 60","plasminogen like B1","DAZ associated protein 2","collagen and calcium binding EGF domains 1","selenoprotein F","transmembrane protein 121B","zinc finger protein 623","EPH receptor A10","coiled-coil domain containing 125","REC114 meiotic recombination protein","bolA family member 2","BCL6 corepressor","JRK like","ciliary associated calcium binding coiled-coil 1","guanylate binding protein family member 6","KIAA2026","ovochymase 2 (gene/pseudogene)","four and a half LIM domains 3","olfactory receptor family 56 subfamily A member 4","chromosome 19 open reading frame 71","coiled-coil domain containing 159","receptor interacting serine/threonine kinase 4","nuclear pore complex interacting protein family member A1","splicing factor 3a subunit 3","tripartite motif containing 61","glutamate ionotropic receptor NMDA type subunit 2A","GTF2H2 family member C","ankyrin repeat and SOCS box containing 7","G protein-coupled receptor 132","MX dynamin like GTPase 2","E1A binding protein p400","terminal nucleotidyltransferase 5C","cytochrome c oxidase assembly factor 5","UTP11 small subunit processome component","proteasome assembly chaperone 1","proline rich 14 like","killer cell lectin like receptor C4","serine hydrolase like 2","poly(rC) binding protein 3","SET domain containing 3, actin histidine methyltransferase","TNF alpha induced protein 8 like 3","zinc and ring finger 3","F-box and leucine rich repeat protein 7","transport and golgi organization 2 homolog","H3 clustered histone 13","sideroflexin 4","mitochondrial ribosomal protein L54","zinc finger protein 438","5-hydroxymethylcytosine binding, ES cell specific","C-C motif chemokine receptor 3","DiGeorge syndrome critical region gene 6","golgin A8 family member G","RP1 like 1","chromosome 11 open reading frame 88","zinc finger protein 530","NADH:ubiquinone oxidoreductase subunit B1","kelch like family member 25","tRNA methyltransferase 12 homolog","pregnancy specific beta-1-glycoprotein 9","bone morphogenetic protein 8a","Aly/REF export factor","refilin B","EF-hand domain containing 2","uridine phosphorylase 1","tripartite motif containing 52","LHFPL tetraspan subfamily member 6","CKLF like MARVEL transmembrane domain containing 4","transmembrane protein 50A","achaete-scute family bHLH transcription factor 2","TANK binding kinase 1","chromobox 6","MET transcriptional regulator MACC1","acyl-CoA synthetase medium chain family member 2A","transducin beta like 3","acid phosphatase 7, tartrate resistant (putative)","kringle containing transmembrane protein 1","TRAF interacting protein","checkpoint kinase 2","apoptosis inducing factor mitochondria associated 3","beta-1,3-galactosyltransferase 5","zinc finger protein 703","tubulin alpha 8","elongin A3 family member D","nuclear pore complex interacting protein family member A5","olfactomedin like 1","RNA binding motif protein 12B","C-C motif chemokine receptor 4","lin-9 DREAM MuvB core complex component","BTB domain containing 9","nudix hydrolase 14","ankyrin repeat domain 45","MYCBP associated and testis expressed 1","G protein-coupled receptor 39","family with sequence similarity 3 member B","zinc finger protein 730","IQ motif containing GTPase activating protein 3","transducer of ERBB2, 2","sodium voltage-gated channel alpha subunit 5","ubiquitously transcribed tetratricopeptide repeat containing, Y-linked","steroid receptor associated and regulated protein",null,"tetratricopeptide repeat domain 32","leucine rich repeat containing 55","dynein axonemal heavy chain 2","SH2 domain containing 1A","short chain dehydrogenase/reductase family 42E, member 2","protein kinase X-linked","lysine methyltransferase 5A","smoothelin","protein phosphatase 2C like domain containing 1","cytochrome c oxidase assembly factor 3","neuropeptide B","ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 3","protein tyrosine phosphatase 4A2","actin gamma 1","transmembrane serine protease 2","DENN domain containing 5A","olfactory receptor family 2 subfamily T member 10","diablo IAP-binding mitochondrial protein","VPS33B late endosome and lysosome associated","T-box transcription factor 1","ArfGAP with dual PH domains 2","ubiquinol-cytochrome c reductase, complex III subunit X","family with sequence similarity 120C","eukaryotic translation initiation factor 3 subunit C","nipsnap homolog 1","contactin 2","leucine rich transmembrane and O-methyltransferase domain containing","potassium voltage-gated channel subfamily Q member 3","nuclear receptor 2C2 associated protein","cysteine rich with EGF like domains 2","olfactory receptor family 1 subfamily D member 2","sec1 family domain containing 2","ubiquitin conjugating enzyme E2 F (putative)","G protein-coupled receptor 173","protein phosphatase 1 regulatory inhibitor subunit 2","TSPY like 2","golgin A6 family like 4","phosphoglycolate phosphatase","small nuclear ribonucleoprotein U11/U12 subunit 35","interleukin 1 receptor associated kinase 1","cms1 ribosomal small subunit homolog","protocadherin 9","acyl-CoA thioesterase 1","out at first homolog","aldehyde dehydrogenase 1 family member A3","cerebellar degeneration related protein 1","H2A clustered histone 20","potassium two pore domain channel subfamily K member 12","H2A clustered histone 21","POU class 6 homeobox 1","TM2 domain containing 3","tumor suppressing subtransferable candidate 4","tumor associated calcium signal transducer 2","C-type lectin like 1","SIX homeobox 6","protein kinase D1","coiled-coil serine rich protein 1","zinc finger DHHC-type containing 23","maestro heat like repeat family member 7","SRSF protein kinase 3","slit guidance ligand 3","ephrin A5","H1.5 linker histone, cluster member","spermatogenesis associated 32","plakophilin 3","MAP7 domain containing 2","colony stimulating factor 1","collectin subfamily member 10","phospholipase A2 group VI","mastermind like transcriptional coactivator 2","SS18L1 subunit of BAF chromatin remodeling complex","DNA topoisomerase I mitochondrial","coatomer protein complex subunit beta 2","leucine rich repeat containing 19","THAP domain containing 7","kinetochore associated 1","C-C motif chemokine receptor 10","non-compact myelin associated protein","WD repeat domain 27","thioredoxin reductase 2","C1q and TNF related 8","forkhead box O4","protein tyrosine phosphatase 4A3","transmembrane protein 255B","protein S","NUT midline carcinoma family member 1","HD domain containing 3","ZFP1 zinc finger protein","dual specificity phosphatase 8","suppressor of cytokine signaling 3","SLIT and NTRK like family member 6","lysophosphatidic acid receptor 5","exportin for tRNA","stimulator of interferon response cGAMP interactor 1","phosphodiesterase 4B","stannin","potassium voltage-gated channel subfamily H member 7","neural EGFL like 2","KRAB-A domain containing 2","mediator complex subunit 12","zinc finger protein 93","septin 9","cell division cycle associated 2","APC membrane recruitment protein 1","zinc finger and BTB domain containing 40","H2B clustered histone 21","claudin 6","olfactory receptor family 51 subfamily M member 1","septin 5","eukaryotic translation initiation factor 4E nuclear import factor 1","leucine rich repeat containing 26","serine incorporator 4","renalase, FAD dependent amine oxidase","apolipoprotein B receptor","family with sequence similarity 110 member C","atlastin GTPase 3","NADH:ubiquinone oxidoreductase subunit A12","t-complex-associated-testis-expressed 3","ubiquitin conjugating enzyme E2 G2","spermidine/spermine N1-acetyl transferase like 1","oxysterol binding protein 2","zinc finger and BTB domain containing 7C","apolipoprotein O","transmembrane p24 trafficking protein 9","transmembrane protein 186","short chain dehydrogenase/reductase family 42E, member 1","RNA binding motif protein 33","armadillo repeat containing X-linked 2","BTB domain containing 6","H1.10 linker histone","RNA binding motif protein 43","small ubiquitin like modifier 3","inner mitochondrial membrane peptidase subunit 2","chloride voltage-gated channel Kb","jagged canonical Notch ligand 2","formin like 1","NUT family member 2A","peptidyl-tRNA hydrolase domain containing 1","lipocalin 12","WT1 transcription factor","ZFP90 zinc finger protein","family with sequence similarity 227 member A","mucin 6, oligomeric mucus/gel-forming","nucleolar complex associated 4 homolog","ubiquitin specific peptidase 18","NADH:ubiquinone oxidoreductase subunit A6","cholinergic receptor muscarinic 5","sortilin related VPS10 domain containing receptor 2","transmembrane protein 121","transmembrane protein 106A","SIVA1 apoptosis inducing factor","diacylglycerol O-acyltransferase 1","roundabout guidance receptor 2","adaptor related protein complex 3 subunit mu 1","coagulation factor VIII","5'-nucleotidase, cytosolic IB","carbonic anhydrase 13","U-box domain containing 5","MAF bZIP transcription factor F","BRF1 RNA polymerase III transcription initiation factor subunit","semaphorin 4B","speedy/RINGO cell cycle regulator family member E16","calcium and integrin binding 1","ankyrin repeat and sterile alpha motif domain containing 1B","negative elongation factor complex member A","solute carrier family 24 member 3","EF-hand calcium binding domain 10","fibronectin leucine rich transmembrane protein 2","integrator complex subunit 5","ribosomal protein S27 like","mannosidase endo-alpha like","adenylosuccinate synthase 1","anoctamin 9","Fas associated factor 1","family with sequence similarity 43 member A","NSE3 homolog, SMC5-SMC6 complex component","heat shock transcription factor 1","chromosome 6 open reading frame 120","purine rich element binding protein A","H2B clustered histone 13","inositol polyphosphate-5-phosphatase J","major facilitator superfamily domain containing 6 like","leucine rich repeat containing 37B","DEAD-box helicase 51","NODAL modulator 2","single Ig and TIR domain containing","nuclear receptor binding protein 2","interferon induced transmembrane protein 2","TNF alpha induced protein 2","zinc finger protein 445","piggyBac transposable element derived 2","transcription elongation factor A like 9","RAB11B, member RAS oncogene family","protein arginine methyltransferase 3","glycoprotein Ib platelet subunit alpha","pre-mRNA processing factor 39","peptidylprolyl isomerase like 6","zinc finger protein 74","KIAA0825","UBA like domain containing 2","cerebral dopamine neurotrophic factor","kelch like family member 33","RNA binding motif protein 11","polypeptide N-acetylgalactosaminyltransferase 17","zinc finger and BTB domain containing 37","interleukin 3 receptor subunit alpha","coiled-coil domain containing 137","RANBP2 like and GRIP domain containing 2","ADP ribosylation factor like GTPase 15","chromosome 12 open reading frame 56","cyclin dependent kinase 10","suppressor of cytokine signaling 1","transcobalamin 2","growth arrest specific 2 like 1","ATPase H+ transporting V0 subunit a2","parkin RBR E3 ubiquitin protein ligase","tubulin epsilon and delta complex 1","hepatocyte growth factor-regulated tyrosine kinase substrate","TNF alpha induced protein 8 like 1","olfactory receptor family 2 subfamily V member 1","RAD51 paralog D","olfactory receptor family 7 subfamily A member 17","mitogen-activated protein kinase 11","SP140 nuclear body protein like","mitochondrial ribosomal protein L30","threonyl-tRNA synthetase 3","SET and MYND domain containing 3","methyltransferase like 7A","interferon lambda receptor 1","SH3 domain binding glutamate rich protein","family with sequence similarity 174 member B","zinc finger SWIM-type containing 9","karyopherin subunit alpha 7","transmembrane protein 179B","GPRIN family member 3","keratin 6B","PARP1 binding protein","SH3 and cysteine rich domain 3","receptor tyrosine kinase like orphan receptor 1","mucin 1, cell surface associated","FA core complex associated protein 100","interferon regulatory factor 7","L3MBTL histone methyl-lysine binding protein 1","BRCA1/BRCA2-containing complex subunit 3","synaptic vesicle glycoprotein 2B","family with sequence similarity 131 member C","lamin tail domain containing 2","protein kinase cGMP-dependent 1","nuclear receptor subfamily 2 group F member 2","TLC domain containing 2","limbic system associated membrane protein","AHNAK nucleoprotein 2","olfactomedin like 2A","Sp1 transcription factor","mitochondrial ribosomal protein L40","inka box actin regulator 1","polycomb group ring finger 3","leishmanolysin like peptidase","prolyl 4-hydroxylase subunit beta","proteasome 26S subunit, non-ATPase 13","PBX homeobox 1","NDUFA4 mitochondrial complex associated like 2","SHC adaptor protein 4","ZFP36 ring finger protein like 1","ubiquitin conjugating enzyme E2 L3","bromodomain and WD repeat domain containing 1","premelanosome protein","synapsin III","POU class 3 homeobox 1","snail family transcriptional repressor 3","zinc finger and BTB domain containing 3","MORN repeat containing 5","chromosome 6 open reading frame 201","MYB proto-oncogene like 1","modulator of smoothened","developmentally regulated GTP binding protein 1","ankyrin repeat and FYVE domain containing 1","YTH N6-methyladenosine RNA binding protein 3","zinc finger protein 696","adenosine deaminase RNA specific B2 (inactive)","sarcalumenin","interferon induced protein with tetratricopeptide repeats 1","chromosome X open reading frame 38","ADAMTS like 5","potassium voltage-gated channel interacting protein 4","mortality factor 4 like 1","WD repeat domain 53","DM1 locus, WD repeat containing","solute carrier family 52 member 2","phosphatidylinositol glycan anchor biosynthesis class P","IKAROS family zinc finger 1","phosphate cytidylyltransferase 2, ethanolamine","nuclear pore associated protein 1","B cell receptor associated protein 31","ADP ribosylation factor like GTPase 17A","G protein subunit beta 1 like","dynein axonemal heavy chain 14","coiled-coil domain containing 190","ecotropic viral integration site 2B","nuclear pore complex interacting protein family member B4","zinc finger protein 829","transmembrane serine protease 11B","threonine synthase like 1","tripartite motif containing 69","ATPase H+ transporting V0 subunit c","interferon induced transmembrane protein 1","lysosomal associated membrane protein 1","protein O-mannose kinase","chromosome 16 open reading frame 54","kelch domain containing 8B","SET domain containing 4","patched 1","reticulon 4 receptor like 1","RNA binding region (RNP1, RRM) containing 3","zinc finger protein 267","insulin receptor substrate 2","chromosome 7 open reading frame 61","family with sequence similarity 186 member A","short stature homeobox","BICD cargo adaptor 2","calicin","trimethyllysine hydroxylase, epsilon","SLIT and NTRK like family member 2","polo like kinase 5","RAS p21 protein activator 3","leucine rich repeats and calponin homology domain containing 3","NADH:ubiquinone oxidoreductase subunit A13","zinc finger protein 566","zinc finger protein 529","zinc finger protein 284","deleted in lymphocytic leukemia 7","axin interactor, dorsalization associated","chromosome 15 open reading frame 41","CD300 molecule like family member f","zona pellucida binding protein 2","keratin 5","gamma-secretase activating protein","ATP/GTP binding protein like 4","arginine-fifty homeobox","cytochrome P450 family 2 subfamily R member 1","leucine rich repeat containing 70","ankyrin repeat domain 46","phosphatidylinositol-4-phosphate 5-kinase type 1 gamma","cytochrome P450 family 4 subfamily F member 2","zinc finger and BTB domain containing 6","chromosome 2 open reading frame 76","RNA polymerase III subunit C","WW domain containing oxidoreductase","cytochrome P450 family 4 subfamily Z member 1","coiled-coil domain containing 84","BCL9 like","RNA polymerase I and III subunit D","kinesin family member 18B","zinc and ring finger 1","free fatty acid receptor 4","suppressor APC domain containing 2","EDAR associated death domain","solute carrier family 51 beta subunit","cytochrome P450 family 4 subfamily F member 12","mitochondrial amidoxime reducing component 1","sosondowah ankyrin repeat domain family member B","biogenesis of lysosomal organelles complex 1 subunit 4","zinc finger protein 749","kelch like family member 32","myocardin related transcription factor B","B and T lymphocyte associated","zinc finger protein 17","lysine demethylase 4D","glycerol-3-phosphate acyltransferase 2, mitochondrial","torsin family 3 member A","protein phosphatase 1 catalytic subunit gamma","zinc finger protein 555","PRELI domain containing 2","beta-secretase 1","transmembrane protein 212","solute carrier family 36 member 3","solute carrier family 36 member 2","thrombospondin 2","retinoid X receptor alpha","ankyrin repeat domain 37","nudix hydrolase 17","membrane integral NOTCH2 associated receptor 2","zinc finger protein 75D","cytochrome P450 family 4 subfamily X member 1","keratin 10","golgin A8 family member R","CD300e molecule","coiled-coil domain containing 30","NFKB repressing factor","gliomedin","Fc fragment of IgA receptor","karyopherin subunit alpha 4","zinc finger protein 501","zinc finger protein 197","spermatogenesis associated 12","transmembrane serine protease 12","ribosomal protein S23","G protein subunit gamma 2","butyrophilin subfamily 3 member A2","A-kinase anchoring protein 14","piccolo presynaptic cytomatrix protein","regulator of G protein signaling 7 binding protein","insulin induced gene 1","myelin transcription factor 1 like","zinc finger protein 396","transmembrane protein 222","chloride voltage-gated channel Ka","Rho GTPase activating protein 30","septin 10","family with sequence similarity 86 member B1","cytochrome P450 family 4 subfamily F member 8","cytochrome P450 family 4 subfamily F member 3","SET and MYND domain containing 4","forkhead box D2","G-patch domain containing 8","CEA cell adhesion molecule 19","neurofibromin 2","small integral membrane protein 29","ubiquitin conjugating enzyme E2 H","4-hydroxyphenylpyruvate dioxygenase like","katanin catalytic subunit A1","fibronectin type III and SPRY domain containing 2","ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1","kinesin family member 24","phosphodiesterase 2A","speedy/RINGO cell cycle regulator family member E17","capping protein regulator and myosin 1 linker 3","proline rich 5","ZFP91 zinc finger protein","chromosome 17 open reading frame 58","BCDIN3 domain containing RNA methyltransferase","cytochrome P450 family 27 subfamily C member 1","LYR motif containing 7","cilia and flagella associated protein 73","coiled-coil domain containing 73","BCR activator of RhoGEF and GTPase","olfactory receptor family 10 subfamily H member 1","fascin actin-bundling protein 2, retinal","forkhead box I2","spindlin family member 4","spindlin family member 2B","hyaluronidase 3","V-set and immunoglobulin domain containing 10 like","C-X-C motif chemokine receptor 3","zinc finger protein 397","zinc finger and SCAN domain containing 30","two pore segment channel 1","leukocyte immunoglobulin like receptor B4","TNF receptor superfamily member 4","keratin 16","HEXIM P-TEFb complex subunit 1","keratin 14","TraB domain containing 2A","PDZ domain containing 7","protein O-fucosyltransferase 2","microtubule associated protein tau","ERCC excision repair 6 like, spindle assembly checkpoint helicase","transmembrane protein 17","TNF receptor superfamily member 18","zinc finger DHHC-type containing 17","zinc finger protein 395","zinc activated ion channel","peroxisome proliferator activated receptor alpha","transmembrane protein 232","family with sequence similarity 183 member A","EF-hand calcium binding domain 6","KN motif and ankyrin repeat domains 3","Rh blood group D antigen","espin","pancreatic lipase related protein 1","peptidyl-tRNA hydrolase 1 homolog","G protein-coupled receptor 141","cytochrome P450 family 4 subfamily A member 11","transmembrane protein 216","ribosomal protein S19 binding protein 1","transmembrane serine protease 11A","transmembrane protein 262","chromosome 3 open reading frame 70","TEA domain transcription factor 1","phospholipase C delta 1","ectonucleoside triphosphate diphosphohydrolase 5 (inactive)","melanocyte inducing transcription factor","HEAT repeat containing 4","nucleosome assembly protein 1 like 1","leukocyte immunoglobulin like receptor A5","C-X9-C motif containing 1","slit guidance ligand 1","LY6/PLAUR domain containing 6","aldo-keto reductase family 1 member C1","spermatogenesis associated 21","ring finger protein 220","angiopoietin like 5","shootin 1","uncharacterized LOC730098","zinc finger protein 546","TSPY like 4","metallothionein 1X","glucosaminyl (N-acetyl) transferase 1","SEC14 and spectrin domain containing 1","formin binding protein 1","dynein cytoplasmic 2 heavy chain 1","keratin 12","basal cell adhesion molecule (Lutheran blood group)","round spermatid basic protein 1 like","WD repeat domain 86","erythropoietin receptor","family with sequence similarity 9 member C","cell death inducing DFFA like effector c","DCC netrin 1 receptor","TATA-box binding protein associated factor 9b","protocadherin beta 13","membrane associated guanylate kinase, WW and PDZ domain containing 2","leucine zipper protein 2","calcineurin like EF-hand protein 1","formyl peptide receptor 3","potassium inwardly rectifying channel subfamily J member 11","cadherin related family member 4","collagen type IV alpha 1 chain","chromosome 12 open reading frame 74","gap junction protein alpha 4","prothymosin alpha","heat shock protein family A (Hsp70) member 14","ATPase 13A5","sirtuin 7","proline rich 27","intraflagellar transport 140","toll like receptor 5","ubiquitin specific peptidase 7","NHL repeat containing E3 ubiquitin protein ligase 1","developmental pluripotency associated 3","pleckstrin homology domain containing N1","zinc finger protein 385C","MAGE family member H1","tet methylcytosine dioxygenase 3","zinc finger protein 286A","ISG15 ubiquitin like modifier","exonuclease 3'-5' domain containing 3","chromosome 17 open reading frame 97","zinc finger with KRAB and SCAN domains 4","RANBP2 like and GRIP domain containing 1","dehydrogenase/reductase 4 like 2","PPARGC1 and ESRR induced regulator, muscle 1","vimentin type intermediate filament associated coiled-coil protein","hyaluronan and proteoglycan link protein 4","ELKS/RAB6-interacting/CAST family member 2","beta 3-glucosyltransferase","sprouty RTK signaling antagonist 4","transient receptor potential cation channel subfamily V member 2","chromosome 2 open reading frame 88","transmembrane protein 203","kelch repeat and BTB domain containing 12","thrombospondin type 1 domain containing 4","DnaJ heat shock protein family (Hsp40) member B13","gamma-aminobutyric acid type A receptor delta subunit","transcription elongation factor A1","non-homologous end joining factor 1","FA complementation group A","SECIS binding protein 2","chromosome 9 open reading frame 153","SSX family member 7","alcohol dehydrogenase 1A (class I), alpha polypeptide","semaphorin 4D","dynein axonemal heavy chain 17","microspherule protein 1","FA complementation group M","zinc finger protein 70","caspase recruitment domain family member 9","platelet endothelial aggregation receptor 1","ZFP69 zinc finger protein B","ZFP69 zinc finger protein","transmembrane protein 220","H1.2 linker histone, cluster member","phospholipid scramblase 3","eukaryotic translation initiation factor 4E binding protein 1","olfactory receptor family 6 subfamily C member 75","coiled-coil domain containing 157","family with sequence similarity 122A","paralemmin 3","FYN binding protein 2","leucine rich repeat containing 74B","deleted in malignant brain tumors 1","C-type lectin domain containing 17A","low density lipoprotein receptor class A domain containing 2","chromosome 2 open reading frame 66","cysteine and histidine rich 1","collagen type XIV alpha 1 chain","delta/notch like EGF repeat containing","kelch like family member 17","phospholipase A2 group IIC","zinc finger and SCAN domain containing 23","Ras and Rab interactor like","chromosome 17 open reading frame 99","olfactory receptor family 7 subfamily D member 2","tumor protein p63 regulated 1","MORN repeat containing 2","ubiquilin 2","Rab interacting lysosomal protein like 1","chromosome 19 open reading frame 67","zinc finger protein 490","NACHT and WD repeat domain containing 1","ADP ribosylation factor like GTPase 4C","transmembrane protein 221","triggering receptor expressed on myeloid cells like 4","RAB42, member RAS oncogene family","Wnt family member 7B","chromosome 11 open reading frame 95","G protein-coupled receptor 89B","eyes shut homolog","chromosome 6 open reading frame 132","olfactory receptor family 2 subfamily AG member 2","mitogen-activated protein kinase 12","NUT family member 2G","collagen type IV alpha 5 chain","agrin","NHS actin remodeling regulator","family with sequence similarity 166 member A","transmembrane protein with metallophosphoesterase domain","zinc finger protein 626","HEPACAM family member 2","smoothelin like 2","zinc finger CCCH-type containing 6","late endosomal/lysosomal adaptor, MAPK and MTOR activator 4","protein kinase cAMP-dependent type I regulatory subunit beta","NUT family member 2B","natural killer cell cytotoxicity receptor 3 ligand 1","defective in cullin neddylation 1 domain containing 3","zinc finger protein 793","tubulin beta 4B class IVb","ArfGAP with GTPase domain, ankyrin repeat and PH domain 4","COMM domain containing 6","interleukin 17 receptor E like","hydroxylysine kinase","olfactory receptor family 7 subfamily A member 5","chromosome 15 open reading frame 62","RUN and FYVE domain containing 4","zinc finger protein 383","hes family bHLH transcription factor 4","zinc finger protein 669","PEAK family member 3","centromere protein P","phospholipid scramblase 1","chromosome 3 open reading frame 62","enolase 4","zinc finger protein 559","SH3 domain binding kinase 1","solute carrier family 38 member 3","general transcription factor IIF subunit 2","family with sequence similarity 92 member A","focadhesin","proline rich 19","zona pellucida glycoprotein 3","Janus kinase and microtubule interacting protein 3","programmed cell death 1","Tctex1 domain containing 4","selectin L","CHM Rab escort protein","biogenesis of lysosomal organelles complex 1 subunit 5","ceramide kinase like","immediate early response 5 like","H2A.X variant histone","chromosome 19 open reading frame 54","lactase like","NCCRP1, F-box associated domain containing","family with sequence similarity 83 member G","cilia and flagella associated protein 77","serine and arginine rich splicing factor 10","hemoglobin subunit alpha 2","dual specificity phosphatase 28","coiled-coil domain containing 9B","NBR1 autophagy cargo receptor","olfactory receptor family 2 subfamily G member 6","Ral GTPase activating protein catalytic alpha subunit 2","NADPH dependent diflavin oxidoreductase 1","cilia and flagella associated protein 54","CLN3 lysosomal/endosomal transmembrane protein, battenin","N-acylsphingosine amidohydrolase 2","small ubiquitin like modifier 2","nanos C2HC-type zinc finger 1","golgin A8 family member M","zinc finger protein 177","retrotransposon Gag like 6","dihydropyrimidine dehydrogenase","S100 calcium binding protein A16","protein prenyltransferase alpha subunit repeat containing 1","coiled-coil and C2 domain containing 2B","stabilizer of axonemal microtubules 2","Rh blood group CcEe antigens","indoleamine 2,3-dioxygenase 2","parvin beta","solute carrier family 4 member 5","uroporphyrinogen III synthase","zinc finger DHHC-type containing 9","ZBED6 C-terminal like","small integral membrane protein 15","von Willebrand factor C domain containing 2","family with sequence similarity 221 member A","transmembrane protein 120B","fibrous sheath interacting protein 2","RNA binding motif protein 34","NADPH oxidase activator 1","transmembrane protein 198","BCL2 like 15","sprouty related EVH1 domain containing 3","placenta expressed transcript 1","adrenoceptor beta 3","proline and arginine rich end leucine rich repeat protein","zinc finger protein 548","metal regulatory transcription factor 1","shisa family member 6","transmembrane protein 201","NHL repeat containing 3","sentan, cilia apical structure protein","zinc finger DHHC-type containing 11","calcium homeostasis modulator family member 6","cannabinoid receptor 2","SLX4 structure-specific endonuclease subunit","ectonucleoside triphosphate diphosphohydrolase 8","ribosomal protein L14","zinc finger protein 563","transmembrane channel like 3","Fas binding factor 1","astacin like metalloendopeptidase","MSL complex subunit 1","putative uncharacterized protein LOC400499","leucine rich repeat kinase 2","gap junction protein beta 3","inhibitory synaptic factor 2A","tRNA methyltransferase 2 homolog B","3-hydroxyacyl-CoA dehydratase 4","cilia and flagella associated protein 126","family with sequence similarity 120A opposite strand","urotensin 2B","chromosome 9 open reading frame 152","NOC2 like nucleolar associated transcriptional repressor","Myb/SANT DNA binding domain containing 1","arylacetamide deacetylase like 3","negative elongation factor complex member B","zinc finger protein 292","potassium channel tetramerization domain containing 21","suprabasin","adenosine deaminase tRNA specific 2","zinc finger protein 567","NDUFA4 mitochondrial complex associated","ankyrin repeat and death domain containing 1B","alkB homolog 2, alpha-ketoglutarate dependent dioxygenase","ring finger protein, transmembrane 1","ring finger protein 222","reelin","family with sequence similarity 111 member B","apolipoprotein D","H1.0 linker histone","lipopolysaccharide induced TNF factor","transmembrane protein 120A","AT-rich interaction domain 2","splicing factor 3b subunit 3","serine protease 48","biogenesis of lysosomal organelles complex 1 subunit 3","Sp6 transcription factor","placenta associated 9","NFKB activating protein like","claudin 4","zinc finger protein 573","family with sequence similarity 47 member E","Jupiter microtubule associated homolog 1","zinc finger protein 527","S100 calcium binding protein A13","zinc finger protein 33A","zinc finger protein 600","KIAA1107","monoamine oxidase A","chromosome 15 open reading frame 61","nuclear GTPase, germinal center associated","TSPY like 1","proline rich nuclear receptor coactivator 2","aspartate rich 1","gap junction protein beta 5","fragile histidine triad diadenosine triphosphatase","zinc finger with KRAB and SCAN domains 3","ribosomal RNA processing 7 homolog A","lin-54 DREAM MuvB core complex component","family with sequence similarity 53 member B","S100 calcium binding protein A14","kazrin, periplakin interacting protein","solute carrier family 35 member E2B","TOG array regulator of axonemal microtubules 2","nuclear envelope integral membrane protein 2","ALG1 chitobiosyldiphosphodolichol beta-mannosyltransferase like","KIAA0408","G1 to S phase transition 2","chromosome 8 open reading frame 76","C-X-C motif chemokine ligand 17","OTU deubiquitinase 6A","high mobility group box 1","SH2 domain containing 5","natural cytotoxicity triggering receptor 1","Ras association domain family member 10","gap junction protein beta 4","biogenesis of lysosomal organelles complex 1 subunit 2","synaptonemal complex protein 2","zinc finger protein 724","interleukin 1 receptor accessory protein","protein tyrosine phosphatase receptor type T","paired box 5","zinc finger protein 676","zinc finger protein 699","tudor domain containing 7","coiled-coil domain containing 189","olfactory receptor family 8 subfamily A member 1","KIAA0895 like","major histocompatibility complex, class II, DR beta 1","myelin transcription factor 1","serpin family A member 3","aldo-keto reductase family 1 member C3","spermatogenesis associated serine rich 2 like","zinc finger protein 250","WD repeat, sterile alpha motif and U-box domain containing 1","zinc finger protein 79","S100 calcium binding protein A4","pleckstrin homology and RhoGEF domain containing G4","FAT atypical cadherin 4","kinesin family member 19","zinc finger protein 681","acyl-CoA dehydrogenase short/branched chain","serine/threonine kinase 40","olfactory receptor family 10 subfamily J member 1","transmembrane protein 63A","semaphorin 4A","M-phase phosphoprotein 8","growth regulating estrogen receptor binding 1","signal regulatory protein beta 2","zinc finger protein 766","ryanodine receptor 1","SLIT-ROBO Rho GTPase activating protein 3","family with sequence similarity 217 member B","tubulin beta class I","ligand dependent nuclear receptor corepressor","SPT5 homolog, DSIF elongation factor subunit","X-prolyl aminopeptidase 3","olfactory receptor family 2 subfamily C member 3","zinc finger protein 107","peptidylprolyl isomerase A","zinc finger protein 471","zinc finger protein 836","zinc finger protein 493","GTF2I repeat domain containing 2","glutamate metabotropic receptor 7","SPT3 homolog, SAGA and STAGA complex component","NGG1 interacting factor 3 like 1","ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1","isoleucyl-tRNA synthetase 1","major facilitator superfamily domain containing 14C","POM121 transmembrane nucleoporin","zinc finger and BTB domain containing 44","GTPase, IMAP family member 5","serine/threonine kinase 31","chorionic gonadotropin subunit beta 7","neuroligin 3","olfactory receptor family 8 subfamily D member 1","alcohol dehydrogenase 7 (class IV), mu or sigma polypeptide","zinc finger with KRAB and SCAN domains 7","CD55 molecule (Cromer blood group)","zinc finger protein 565","netrin G2","ELAV like RNA binding protein 3","WD repeat domain 5","lon peptidase 1, mitochondrial","transformation/transcription domain associated protein","SLIT-ROBO Rho GTPase activating protein 2B","fucosyltransferase 4","ankyrin repeat and SOCS box containing 13","zinc finger protein 34","zinc finger protein 781","zinc finger protein 140","inhibitor of CDK, cyclin A1 interacting protein 1","zinc finger protein 774","protein tyrosine phosphatase non-receptor type 1","Enah/Vasp-like","thioesterase superfamily member 5","EPH receptor B4","zinc finger protein 765","zinc finger protein 124","X-ray repair cross complementing 6","S100 calcium binding protein A5","chromosome 20 open reading frame 204","protein phosphatase 1 regulatory subunit 26","TSC22 domain family member 2","nuclear pore complex interacting protein family member B15","zinc finger protein 569","armadillo repeat containing X-linked 4","yrdC N6-threonylcarbamoyltransferase domain containing","zinc finger protein 777","phosphoinositide-3-kinase regulatory subunit 4","zinc finger protein 775","zinc finger protein 605","trafficking protein particle complex 2","myosin light chain 6B","zinc finger protein 799","siah E3 ubiquitin protein ligase 1","chromosome 20 open reading frame 96","estrogen related receptor gamma","importin 4","nuclear receptor corepressor 2","sulfotransferase family 1A member 1","ADP ribosylation factor like GTPase 9","pre-mRNA processing factor 40 homolog A","ganglioside induced differentiation associated protein 2","transcription elongation factor A like 3","anaphase promoting complex subunit 7","thiamin pyrophosphokinase 1","solute carrier family 6 member 9","actin filament associated protein 1","nascent polypeptide associated complex subunit alpha","myosin XVIIIA","serine palmitoyltransferase small subunit B","BLOC-1 related complex subunit 6","mannosidase alpha class 2A member 2","membrane metalloendopeptidase","family with sequence similarity 72 member A","coiled-coil domain containing 196","calcium voltage-gated channel subunit alpha1 H","sulfatase 2","laminin subunit alpha 2","plexin B2","adherens junctions associated protein 1","X-ray repair cross complementing 2","myosin VI","myocardin related transcription factor A","histone deacetylase 2","zinc finger protein 782","solute carrier family 22 member 25","zinc finger protein 846","matrix metallopeptidase 1","alcohol dehydrogenase 1B (class I), beta polypeptide","transcription factor 4","WNK lysine deficient protein kinase 3","succinate dehydrogenase complex assembly factor 3","histamine receptor H1","RAB, member RAS oncogene family like 6","zinc finger protein 136","zinc finger with KRAB and SCAN domains 5","zinc finger protein 502","trafficking protein particle complex 4","tetratricopeptide repeat domain 30B","tectonin beta-propeller repeat containing 2","toll like receptor 7","family with sequence similarity 180 member B","ZFP62 zinc finger protein","ERI1 exoribonuclease family member 2","translocase of outer mitochondrial membrane 7","hematopoietic SH2 domain containing","transient receptor potential cation channel subfamily V member 1","zinc finger protein 33B","zinc finger protein 512B","archaelysin family metallopeptidase 2","zinc finger protein 431","neurofibromin 1","vitamin K epoxide reductase complex subunit 1 like 1","zinc finger protein 418","death associated protein kinase 1","major histocompatibility complex, class II, DQ alpha 1","collagen type XXVII alpha 1 chain","GM2 ganglioside activator","H2A clustered histone 13","S100 calcium binding protein A2","zinc finger protein 700","CD47 molecule","olfactory receptor family 52 subfamily K member 1","TLE family member 1, transcriptional corepressor","mastermind like transcriptional coactivator 3","H2A clustered histone 11","striatin 3","cholinergic receptor nicotinic gamma subunit","zinc finger and SCAN domain containing 16","multivesicular body subunit 12B","inflammation and lipid regulator with UBA-like and NBR1-like domains",null,"olfactory receptor family 11 subfamily G member 2","POTE ankyrin domain family member I","adenosine deaminase","AT-rich interaction domain 5A","prostate and testis expressed 2","PTC7 protein phosphatase homolog","keratin 39","NHL repeat containing 2","H2A clustered histone 7","ZFP28 zinc finger protein","KIAA1211 like","COBW domain containing 3","sodium voltage-gated channel alpha subunit 8","laminin subunit beta 3","H2B.U histone 1","karyopherin subunit alpha 5","ankyrin repeat domain 36B","Rho guanine nucleotide exchange factor 12","hydroxycarboxylic acid receptor 1","PDZ and LIM domain 7","filamin A","SLIT-ROBO Rho GTPase activating protein 1","family with sequence similarity 3 member C","NOP9 nucleolar protein","solute carrier family 39 member 10","caspase 4","adaptor related protein complex 2 subunit alpha 1","zinc finger protein 585A","fucosyltransferase 11","annexin A4","L antigen family member 3","WD repeat domain 5B","nuclear pore complex interacting protein family member B9","WD repeat domain 45","methyltransferase like 9","zinc finger protein 138","zinc finger protein 429","zinc finger protein 470","SERTA domain containing 1","zinc finger protein 100","chromosome X open reading frame 40B","zinc finger protein 398","zinc finger and SCAN domain containing 25","annexin A6","zinc finger protein 441","glia maturation factor beta","sialic acid binding Ig like lectin 15","zinc finger protein 420","zinc finger protein 763","zinc finger MYM-type containing 1","death domain containing 1","H4 clustered histone 3","zinc finger and SCAN domain containing 26","MAF bZIP transcription factor G","arrestin domain containing 1","KIAA1671","insulin like growth factor 2 receptor","galactose-3-O-sulfotransferase 4","dynein cytoplasmic 1 heavy chain 1","solute carrier family 6 member 17","poly(rC) binding protein 2","zinc finger CCCH-type and G-patch domain containing","solute carrier family 25 member 29","post-GPI attachment to proteins inositol deacylase 1","SRC proto-oncogene, non-receptor tyrosine kinase","zinc finger protein 682","olfactory receptor family 8 subfamily B member 8","zinc finger protein 772","zinc finger protein 257","pecanex 3","ADAM metallopeptidase domain 32","acyl-CoA synthetase long chain family member 5","leucine rich repeat containing 8 VRAC subunit B","ATP binding cassette subfamily B member 8","H3 clustered histone 12","staphylococcal nuclease and tudor domain containing 1","zinc finger protein 785","sulfotransferase family 1A member 2","NIMA related kinase 5","proteasome 26S subunit, non-ATPase 12","piwi like RNA-mediated gene silencing 2","nucleolar protein 4 like","cysteine rich tail 1","solute carrier family 22 member 4","ectonucleoside triphosphate diphosphohydrolase 4","C1D nuclear receptor corepressor","TBC1 domain family member 9B","H4 clustered histone 11","family with sequence similarity 110 member D","serpin family A member 1","tryptase beta 2 (gene/pseudogene)","KN motif and ankyrin repeat domains 2","chromosome 6 open reading frame 141","general transcription factor IIE subunit 2","interleukin 27","RAD54 homolog B","zinc finger protein 165","synaptic Ras GTPase activating protein 1","fat storage inducing transmembrane protein 2","BLM RecQ like helicase","zinc finger protein 720","DNA damage inducible 1 homolog 2","supervillin","tripartite motif containing 33","LDL receptor related protein 10","pellino E3 ubiquitin protein ligase 1","zinc finger protein 655","mitochondrial ribosomal protein L21","LY6/PLAUR domain containing 2","UDP-N-acetylglucosamine pyrophosphorylase 1 like 1","F-box and leucine rich repeat protein 22","zinc finger protein 786","zinc finger protein 517","zinc finger protein 675","solute carrier family 22 member 5","dishevelled binding antagonist of beta catenin 3","adenosine deaminase RNA specific B1","zinc finger protein 860","huntingtin","complement C5a receptor 1","cytochrome P450 family 2 subfamily B member 6","H3 clustered histone 4","dachsous cadherin-related 2","ventricular zone expressed PH domain containing 1","sedoheptulokinase","intracisternal A particle-promoted polypeptide","mitogen-activated protein kinase kinase kinase 5","cytochrome P450 family 2 subfamily F member 1","glutathione S-transferase kappa 1","heterogeneous nuclear ribonucleoprotein A/B","olfactory receptor family 2 subfamily L member 5","stathmin 3","platelet derived growth factor subunit A","glycophorin E (MNS blood group)","sialophorin","zinc finger protein 695","protocadherin beta 11","zinc finger protein 628","solute carrier family 2 member 10","zinc finger protein 665","ribosome production factor 2 homolog","solute carrier family 28 member 3","family with sequence similarity 177 member B","mindbomb E3 ubiquitin protein ligase 2","olfactory receptor family 6 subfamily Y member 1","myosin VA","granzyme M","autophagy related 7","signal induced proliferation associated 1 like 1","tetratricopeptide repeat domain 30A","RAB40C, member RAS oncogene family","phosphatidylinositol glycan anchor biosynthesis class N","collagen type IV alpha 6 chain","zinc finger protein 624","HERV-H LTR-associating 3","TOP1 binding arginine/serine rich protein","beta-carotene oxygenase 2","potassium calcium-activated channel subfamily M regulatory beta subunit 2","ectonucleoside triphosphate diphosphohydrolase 6","diencephalon/mesencephalon homeobox 1","ectonucleotide pyrophosphatase/phosphodiesterase 1","coiled-coil domain containing 154","fatty acyl-CoA reductase 1","ciliogenesis and planar polarity effector 1","zinc finger protein 841","microfibril associated protein 5","zinc finger protein 615","chromosome X open reading frame 40A","CDC42 small effector 1","macrophage expressed 1","serpin family B member 2","dipeptidyl peptidase 4","serpin family B member 13","programmed cell death 1 ligand 2","zinc finger protein 433","dynein axonemal heavy chain 10","spectrin alpha, non-erythrocytic 1","neuromedin B","parvin alpha","kelch like family member 14","family with sequence similarity 114 member A1","ribulose-5-phosphate-3-epimerase","zinc finger protein 460","complement C3b/C4b receptor 1 like","PHD finger protein 2","ribosomal protein S26","chromosome 14 open reading frame 178","prosaposin","S100 calcium binding protein A10","cilia and flagella associated protein 43","LHFPL tetraspan subfamily member 5","ribosomal protein L37a","thioredoxin reductase 3","complement factor D","sperm-tail PG-rich repeat containing 3","minichromosome maintenance complex binding protein","essential meiotic structure-specific endonuclease subunit 2","kelch domain containing 1","zinc finger protein 81","TATA-box binding protein associated factor 13","zinc finger protein 780A","ATPase family AAA domain containing 3A","family with sequence similarity 118 member B","zinc finger protein 461","coiled-coil domain containing 180","solute carrier family 9 member A8","occludin","cilia and flagella associated protein 299","H4 histone 16","cytochrome P450 family 2 subfamily A member 13","zinc finger protein 181","olfactory receptor family 8 subfamily G member 1 (gene/pseudogene)","inka box actin regulator 2","zinc finger protein 44","glycosylphosphatidylinositol anchor attachment 1","ADAMTS like 2","small glutamine rich tetratricopeptide repeat containing beta","zinc finger protein 790","proline rich protein BstNI subfamily 3","family with sequence similarity 49 member A","myosin IC","NFKB inhibitor interacting Ras like 1","meiosis/spermiogenesis associated 1","kinesin family member 13B","alcohol dehydrogenase 5 (class III), chi polypeptide","H2B clustered histone 12","SPG7 matrix AAA peptidase subunit, paraplegin","hornerin","zinc finger protein 677","endoplasmic reticulum oxidoreductase 1 alpha","zinc finger protein 823","zinc finger protein 311","zinc finger protein 347","phospholipase C gamma 2","FCH and double SH3 domains 1","zinc finger protein 71","arylacetamide deacetylase like 2","S100 calcium binding protein A6","ribosomal protein L12","dynamin 3","zinc finger protein 121","myelin protein zero like 1","vacuolar protein sorting 13 homolog A","myelin basic protein","A-kinase anchoring protein 17A","golgin A6 family like 9","leucine, glutamate and lysine rich 1","chromosome 1 open reading frame 122","C-type lectin domain containing 9A","nucleolar protein 8","interleukin 1 receptor associated kinase 4","coiled-coil domain containing 151","mitochondrial ribosomal protein L42","ectonucleoside triphosphate diphosphohydrolase 7","Fc fragment of IgG receptor Ib","zinc finger protein 335","ribosomal protein S4 X-linked","zinc finger protein 273","zinc finger protein 84","MAK16 homolog","zinc finger protein 667","signal regulatory protein alpha","G protein-coupled receptor kinase 6","DNA primase subunit 1","membrane associated ring-CH-type finger 5","POTE ankyrin domain family member H","nuclear pore complex interacting protein family, member B13","aldo-keto reductase family 1 member B10","sulfotransferase family 1C member 4","cytochrome P450 family 2 subfamily A member 7","zinc finger and BTB domain containing 14","CD2 associated protein","nucleoporin 62 C-terminal like","SFI1 centrin binding protein","zinc finger protein 649","alcohol dehydrogenase 4 (class II), pi polypeptide","olfactory receptor family 2 subfamily T member 6","zinc finger protein 248","torsin family 4 member A","lysophosphatidic acid receptor 1","myoglobin","olfactory receptor family 2 subfamily L member 3","3-hydroxyisobutyryl-CoA hydrolase","zinc finger protein 544","transmembrane protein 229B","sosondowah ankyrin repeat domain family member C","zinc finger protein 770","nuclear pore complex interacting protein family member B6","high mobility group nucleosome binding domain 5","MIER1 transcriptional regulator","mannosidase alpha class 1A member 2","small VCP interacting protein","zinc finger protein 251","DDRGK domain containing 1","transcription factor Dp-1","C-type lectin domain family 4 member C","zinc finger protein 607","BPI fold containing family A member 1","zinc finger protein 334","hydroxysteroid 17-beta dehydrogenase 11","SZT2 subunit of KICSTOR complex","sulfotransferase family 1C member 2","zinc finger X-linked duplicated A","ribosomal protein S6 kinase like 1","calcium voltage-gated channel subunit alpha1 E","glutamine rich 1","colony stimulating factor 2 receptor alpha subunit","DEAD-box helicase 42","ribosomal protein L23a","solute carrier family 29 member 3","serine/threonine/tyrosine interacting protein","ubiquitin like 5","helicase with zinc finger","transmembrane protein 116","uridine-cytidine kinase 1 like 1","caspase recruitment domain family member 11","zinc finger protein 485","SDA1 domain containing 1","zinc finger with KRAB and SCAN domains 8","PH domain containing endocytic trafficking adaptor 1","HYLS1 centriolar and ciliogenesis associated","zinc finger protein 442","zinc finger protein 813","Pim-3 proto-oncogene, serine/threonine kinase","guided entry of tail-anchored proteins factor 3, ATPase","aspartate beta-hydroxylase","sprouty related EVH1 domain containing 2","WW domain containing E3 ubiquitin protein ligase 2","glutamine--fructose-6-phosphate transaminase 1","UV radiation resistance associated","zinc finger protein 26","intersectin 2","neurotrophic receptor tyrosine kinase 1","O-GlcNAcase","metallothionein 1F","TRPM8 channel associated factor 1","zinc finger protein 69","thioredoxin reductase 1","NOTCH regulated ankyrin repeat protein","zinc finger protein 583","zinc finger protein 568","zinc finger X-linked duplicated B","zinc finger protein 480","zinc finger protein 587","tropomyosin 2","receptor transporter protein 2","SH3 domain binding glutamate rich protein like 2","zinc finger protein 808","ankyrin repeat domain 35","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 6","YTH N6-methyladenosine RNA binding protein 2","translation machinery associated 16 homolog","major histocompatibility complex, class II, DR beta 5","atlastin GTPase 1","cyclic nucleotide gated channel subunit alpha 1","MAF bZIP transcription factor K","armadillo like helical domain containing 1","zinc finger protein 43","GPN-loop GTPase 1","phospholamban","C2 calcium dependent domain containing 4A","zinc finger protein 28","zinc finger protein 511","chromosome 20 open reading frame 203","zinc finger protein 627","potassium channel regulator","WD repeat and HMG-box DNA binding protein 1","zinc finger protein 789","catenin delta 1","DExD-box helicase 39B","solute carrier family 34 member 3","retinal degeneration 3, GUCY2D regulator","SH2 domain containing 1B","activity regulated cytoskeleton associated protein","nudix hydrolase 16","tousled like kinase 1","LPS responsive beige-like anchor protein","bromodomain adjacent to zinc finger domain 1A","aldo-keto reductase family 1 member C4","COP9 signalosome subunit 8","coiled-coil domain containing 69","MDM4 regulator of p53","ryanodine receptor 2","kelch like family member 9","family with sequence similarity 3 member D","nuclear receptor coactivator 6","serine/threonine kinase 39","tyrosine aminotransferase","chromosome 6 open reading frame 89","calmodulin 1","lipoprotein(a)","TAFA chemokine like family member 2","tetratricopeptide repeat domain 37","tumor suppressor candidate 1","3'-phosphoadenosine 5'-phosphosulfate synthase 2","solute carrier family 9 member A6","FANCD2 and FANCI associated nuclease 1","ATP binding cassette subfamily A member 4","eukaryotic translation initiation factor 1A Y-linked","NADH dehydrogenase, subunit 6 (complex I)","importin 9","centrosomal protein 290","cytochrome c oxidase subunit II","glycosylated lysosomal membrane protein","TOG array regulator of axonemal microtubules 1","delta like canonical Notch ligand 1","ankyrin repeat domain 13B","enoyl-CoA delta isomerase 2","unc-13 homolog B","testis expressed 45","cytochrome b","LIM domain binding 1","protein phosphatase 1 regulatory inhibitor subunit 14C","CTR9 homolog, Paf1/RNA polymerase II complex component","SPARC related modular calcium binding 1","coagulation factor V","methionine sulfoxide reductase B1","zinc finger protein 652","SMAD specific E3 ubiquitin protein ligase 1","solute carrier family 5 member 3","G-patch domain containing 3","CDC42 binding protein kinase beta","plexin B3","ribosomal protein L10a","collagen beta(1-O)galactosyltransferase 2","EPS8 like 3","EGF like domain multiple 6","MTND2","synaptonemal complex protein 1","APC down-regulated 1 like","RCSD domain containing 1","Ras association domain family member 9","family with sequence similarity 169 member A","zinc finger protein 830","glutamate ionotropic receptor NMDA type subunit 3A","NADH dehydrogenase, subunit 5 (complex I)","mucin 2, oligomeric mucus/gel-forming","CCR4-NOT transcription complex subunit 7","transmembrane protein 184B","mechanistic target of rapamycin kinase","secretory carrier membrane protein 5","alpha kinase 2","leucine rich repeats and immunoglobulin like domains 2","cytochrome c oxidase subunit I","purine nucleoside phosphorylase","paired box 9","leucine rich repeat containing 10","glycerol kinase","forkhead box J3","zinc finger protein 358","SFT2 domain containing 1","CD247 molecule","glutamate metabotropic receptor 3","chromosome alignment maintaining phosphoprotein 1","inositol polyphosphate-5-phosphatase F","Rho GTPase activating protein 11A","succinate receptor 1","high mobility group nucleosomal binding domain 2","selenoprotein M","ubiquitin conjugating enzyme E2 J1","gap junction protein gamma 2","OPA1 mitochondrial dynamin like GTPase","DENN domain containing 4B","ryanodine receptor 3","zinc finger protein 277","NADH dehydrogenase, subunit 3 (complex I)","KTI12 chromatin associated homolog","selenoprotein T","thymocyte selection associated high mobility group box","carboxylesterase 1","CD3e molecule","RUN and SH3 domain containing 2","FIC domain containing","oligosaccharyltransferase complex non-catalytic subunit","R3H domain containing 4","tRNA splicing endonuclease subunit 15","listerin E3 ubiquitin protein ligase 1","RUN domain containing 1","coiled-coil domain containing 152","serine/threonine kinase like domain containing 1","G protein-coupled receptor kinase 5","tRNA-yW synthesizing protein 1 homolog","DDB1 and CUL4 associated factor 12","Scm like with four mbt domains 2","ITPRIP like 1","NADH dehydrogenase, subunit 4 (complex I)","structural maintenance of chromosomes 5","NADH dehydrogenase, subunit 1 (complex I)","protein arginine methyltransferase 6","shisa family member 4","CLOCK interacting pacemaker","capping actin protein of muscle Z-line subunit alpha 2","ATP synthase F0 subunit 6","DNA topoisomerase I","protein regulator of cytokinesis 1","basic helix-loop-helix family member b9","mitogen-activated protein kinase kinase kinase 3","sterol regulatory element binding transcription factor 2","chromosome 1 open reading frame 174","RasGEF domain family member 1A","SPOUT domain containing methyltransferase 1","ribosomal protein L39","DAZ interacting zinc finger protein 3","KIAA0753","DNA cross-link repair 1A","autophagy related 9A","nitric oxide synthase 1 adaptor protein","adenine phosphoribosyltransferase","G protein-coupled receptor associated sorting protein 1","TBK1 binding protein 1","MAGE family member E1","coiled-coil domain containing 167","cytochrome c oxidase III","ZFP2 zinc finger protein","sosondowah ankyrin repeat domain family member A","L3MBTL histone methyl-lysine binding protein 3","dystrophin","microfibril associated protein 3 like","alpha-N-acetylgalactosaminidase","SMG5 nonsense mediated mRNA decay factor","kinesin family binding protein","transglutaminase 2","armadillo repeat containing X-linked 6","praja ring finger ubiquitin ligase 2","RAR related orphan receptor B","sphingomyelin synthase 1","olfactory receptor family 10 subfamily Z member 1","zinc finger protein 525","inverted formin, FH2 and WH2 domain containing","FAM83H antisense RNA 1 (head to head)",null,"olfactory receptor family 2 subfamily L member 2","EF-hand calcium binding domain 2","cytochrome c oxidase assembly factor COX20","CHM like Rab escort protein","calpain 8","TatD DNase domain containing 3","complement C3b/C4b receptor 1 (Knops blood group)","retinoic acid early transcript 1G","sterile alpha motif domain containing 5","transmembrane epididymal protein 1","epithelial cell transforming 2 like","G protein-coupled receptor 52","Fc fragment of IgG receptor IIIa","shadow of prion protein","family with sequence similarity 229 member B","fibronectin type III and ankyrin repeat domains 1","proline rich 9","small proline rich protein 2E","EEF1A lysine methyltransferase 2","D-aspartate oxidase","phospholipid phosphatase 4","H3 clustered histone 14","H2A clustered histone 18","H2B clustered histone 18","H3 clustered histone 15","RNA binding motif protein 20","chromosome 6 open reading frame 163","GDP dissociation inhibitor 1","protein-L-isoaspartate (D-aspartate) O-methyltransferase domain containing 2","Lck interacting transmembrane adaptor 1","developmental pluripotency associated 5","sterile alpha motif domain containing 13","retrotransposon Gag like 8A","coiled-coil domain containing 160","chromosome 1 open reading frame 141","EF-hand calcium binding domain 7","low density lipoprotein receptor class A domain containing 1","Rhox homeobox family member 2B","zyg-11 family member A, cell cycle regulator","lipase family member N","leucine rich repeat containing 73","forkhead box O6","SYS1 golgi trafficking protein","inositol polyphosphate-5-phosphatase B","neuraminidase 4","MAF bZIP transcription factor B","TRAF3 interacting protein 1","cysteine rich hydrophobic domain 1","GRB10 interacting GYF protein 2","chromosome 2 open reading frame 72","RUN and FYVE domain containing 2","NHS like 2","phosphatase and actin regulator 4","N-acylsphingosine amidohydrolase 2B","ArfGAP with GTPase domain, ankyrin repeat and PH domain 6","translocase of inner mitochondrial membrane 23 homolog B","zinc finger DHHC-type containing 18","transmembrane protein 273","chromosome X open reading frame 65","ArfGAP with GTPase domain, ankyrin repeat and PH domain 9","synaptotagmin 15","macoilin 1","protein tyrosine phosphatase non-receptor type 20","growth differentiation factor 5 opposite strand","zinc finger DBF-type containing 2","thioredoxin domain containing 8","death domain associated protein","bone morphogenetic protein receptor type 2","transcription elongation factor A3","prefoldin subunit 6","ring finger protein 1","hydroxysteroid 17-beta dehydrogenase 8","retinoid X receptor beta","oxidoreductase like domain containing 1","major histocompatibility complex, class II, DO alpha","bromodomain containing 2","major histocompatibility complex, class II, DM alpha","collagen type V alpha 2 chain","proteasome 20S subunit beta 8","transporter 2, ATP binding cassette subfamily B member","spindlin family member 3","negative regulator of P-body association","major histocompatibility complex, class II, DR alpha","collagen type XV alpha 1 chain","notch receptor 4","PBX homeobox 2","advanced glycosylation end-product specific receptor","ring finger protein 5","1-acylglycerol-3-phosphate O-acyltransferase 1","pejvakin","proline rich transmembrane protein 1","FKBP prolyl isomerase like","mitochondrial ribosomal protein L38","small integral membrane protein 5","glutamate rich 2","serine/threonine kinase 19","CD300 molecule like family member d","decapping exoribonuclease","Ski2 like RNA helicase","negative elongation factor complex member E","zinc finger and BTB domain containing 12","succinate dehydrogenase complex subunit D","euchromatic histone lysine methyltransferase 2","layilin","solute carrier family 44 member 4","neuraminidase 1","heat shock protein family A (Hsp70) member 1B","heat shock protein family A (Hsp70) member 1A","heat shock protein family A (Hsp70) member 1 like","LSM2 homolog, U6 small nuclear RNA and mRNA degradation associated","valyl-tRNA synthetase 1","von Willebrand factor A domain containing 7","caspase recruitment domain family member 16","methyl-CpG binding domain protein 5","mutS homolog 5","megakaryocyte and platelet inhibitory receptor G6b","lymphocyte antigen 6 family member G6C","abhydrolase domain containing 16A","lymphocyte antigen 6 family member G5C","casein kinase 2 beta","G-patch domain and ankyrin repeats 1","chromosome 6 open reading frame 47","family with sequence similarity 155 member A","apolipoprotein M","BCL2 associated athanogene 6","proline rich coiled-coil 2A","allograft inflammatory factor 1","leukocyte specific transcript 1","NFKB inhibitor like 1","zinc finger protein 814","MHC class I polypeptide-related sequence B","zinc finger protein 551","MHC class I polypeptide-related sequence A","zinc finger protein 805","major histocompatibility complex, class I, C","POU class 5 homeobox 1","coiled-coil alpha-helical rod protein 1","psoriasis susceptibility 1 candidate 1","mucin 21, cell surface associated","DEAH-box helicase 16","chromosome 6 open reading frame 136","mitochondrial ribosomal protein S18B","protein phosphatase 1 regulatory subunit 10","keratin associated protein 5-11","ATP binding cassette subfamily F member 1","proline rich 3","leukocyte immunoglobulin like receptor B3","discoidin domain receptor tyrosine kinase 1","G protein nucleolar 1 (putative)","major histocompatibility complex, class I, E","tripartite motif containing 39","zinc finger protein 468","zinc finger protein 616","tripartite motif containing 31","ring finger protein 39","protein phosphatase 1 regulatory inhibitor subunit 11","receptor for activated C kinase 1","major histocompatibility complex, class I, G","TBC1 domain family member 8","major histocompatibility complex, class I, F","aspartate dehydrogenase domain containing","myelin oligodendrocyte glycoprotein","olfactory receptor family 2 subfamily H member 2","AKT1 substrate 1","gamma-aminobutyric acid type B receptor subunit 1","chromosome 10 open reading frame 113","olfactory receptor family 11 subfamily A member 1","olfactory receptor family 14 subfamily J member 1","olfactory receptor family 2 subfamily J member 3","chromosome 9 open reading frame 135","tripartite motif containing 27","RAN binding protein 17","inhibitory synaptic factor family member 2B","forkhead box D4 like 5",null,"tetratricopeptide repeat domain 25","mitochondrial ribosomal protein L53","maestro heat like repeat family member 6","ataxin 2","dynactin subunit 1","tectonic family member 1","family with sequence similarity 216 member A","zinc finger and BTB domain containing 48","IGF like family member 2","IGF like family member 4","keratin 40","mitotic spindle organizing protein 1","serine peptidase inhibitor, Kazal type 9","zinc finger protein 155","ubiquinol-cytochrome c reductase complex assembly factor 3","F-box protein 48","family with sequence similarity 221 member B","CD177 molecule","pregnancy specific beta-1-glycoprotein 5","zinc finger family member 783","zinc finger protein 425","leucine rich repeat containing 10B","chromosome 12 open reading frame 73","protocadherin gamma subfamily A, 1","protocadherin alpha 9","protocadherin alpha 5","protocadherin alpha 4","tripartite motif containing 13","spire type actin nucleation factor 2","PKHD1 like 1","schlafen family member 12 like","solute carrier family 35 member B4","galectin 7","synaptonemal complex central element protein 1 like","transmembrane protein 231","family with sequence similarity 71 member F2","chromosome 2 open reading frame 91","cyclin I family member 2","cyclin dependent kinase like 4","chromosome 4 open reading frame 47","triple QxxK/R motif containing","succinate dehydrogenase complex assembly factor 1","presenilin enhancer, gamma-secretase subunit","chromosome 11 open reading frame 91","zinc finger and BTB domain containing 10","chromosome 4 open reading frame 46","secretoglobin family 2B member 2","coiled-coil domain containing 144 family, N-terminal like","leucine rich repeat containing G protein-coupled receptor 4","proteasome 20S subunit beta 10",null,"speedy/RINGO cell cycle regulator family member E2","E2F transcription factor 4","phosphodiesterase 7A","transmembrane protein 170B","mucin 12, cell surface associated","sorting nexin 2","Sin3A associated protein 25","SAP domain containing ribonucleoprotein","adhesion G protein-coupled receptor G1","importin 7","proline rich 13","tectonin beta-propeller repeat containing 1","solute carrier organic anion transporter family member 6A1","inhibitory synaptic factor 1","complement factor I","sterile alpha motif domain containing 9","keratin 6A","CTD nuclear envelope phosphatase 1 regulatory subunit 1","keratin 81","exocyst complex component 3 like 4","ATPase H+ transporting accessory protein 1 like","coiled-coil domain containing 85C","C2 calcium dependent domain containing 4B","ral guanine nucleotide dissociation stimulator like 3","nucleosome assembly protein 1 like 4","thymosin beta 4 X-linked","transmembrane protein 256","carnitine palmitoyltransferase 1B","survival of motor neuron 1, telomeric","small EDRK-rich factor 1A","high mobility group nucleosome binding domain 1","mucin 19, oligomeric","DENN domain containing 6B","eukaryotic translation initiation factor 3 subunit C like","leucine carboxyl methyltransferase 1","major facilitator superfamily domain containing 2B","cysteine rich DPF motif domain containing 1","histatin 3","lin-52 DREAM MuvB core complex component","small integral membrane protein 11A","trans-2,3-enoyl-CoA reductase like","double PHD fingers 3","electron transfer flavoprotein regulatory factor 1","chromosome 17 open reading frame 107","intersectin 1","ITPRIP like 2","DENN domain containing 1C","solute carrier organic anion transporter family member 1B7 (putative)","crystallin zeta like 1","chromosome 5 open reading frame 51","G antigen 1","phospholipid phosphatase 6","killer cell lectin like receptor C2","geminin coiled-coil domain containing","tetratricopeptide repeat domain 23 like","C1q and TNF related 9B","zinc finger protein 316","one cut homeobox 3","chromosome 21 open reading frame 62","RNA binding protein with serine rich domain 1","NYN domain and retroviral integrase containing","DnaJ heat shock protein family (Hsp40) member C19","defensin alpha 1","docking protein 6","Jupiter microtubule associated homolog 2","serpin family B member 11 (gene/pseudogene)","serpin family B member 4","serpin family B member 5","zinc finger DHHC-type containing 11B","cilia and flagella associated protein 99","golgin A8 family member O","transmembrane protein 191C","hemoglobin subunit alpha 1","elongin A2","ATPase phospholipid transporting 10A (putative)","ankyrin repeat and ubiquitin domain containing 1","FOXL2 neighbor","collagen type VI alpha 6 chain","RAB12, member RAS oncogene family","transmembrane protein 200C","major histocompatibility complex, class I, A","3-hydroxyacyl-CoA dehydratase 2","cilia and flagella associated protein 44","CD200 receptor 1 like","leukemia NUP98 fusion partner 1","vestigial like family member 3","tripartite motif containing 71","zinc finger CW-type and PWWP domain containing 2","ankyrin repeat domain 28","collagen like tail subunit of asymmetric acetylcholinesterase","methyltransferase like 6","XK related 4","glutathione peroxidase 3","iodothyronine deiodinase 2","selenoprotein H","iodothyronine deiodinase 1","serine/threonine kinase 38 like","translin","solute carrier family 48 member 1","testis specific serine kinase 1B","proline rich 22","taste 2 receptor member 19","taste 2 receptor member 14","taste 2 receptor member 13","cutaneous T cell lymphoma-associated antigen 1","retrotransposon Gag like 8B","olfactory receptor family 2 subfamily A member 42","ring finger protein 208","NADH dehydrogenase, subunit 4L (complex I)","microtubule associated protein 10","POU class 5 homeobox 1B","zinc finger protein 580","zinc finger protein 611","nucleoporin 62","DENN domain containing 1B","SFT2 domain containing 2","FGFR1 oncogene partner","SR-related CTD associated factor 8","cilia and flagella associated protein 45","zinc finger protein 254","Tctex1 domain containing 2","crystallin gamma S","cysteine rich protein 1","kelch like family member 23","leucine rich repeat and Ig domain containing 4","tripartite motif containing 59","MLLT11 transcription factor 7 cofactor","acid sensing ion channel subunit 3","GTPase, IMAP family member 1",null,"coiled-coil domain containing 183","Rho guanine nucleotide exchange factor 35","olfactory receptor family 2 subfamily F member 1 (gene/pseudogene)","DNL-type zinc finger","SPT4 homolog, DSIF elongation factor subunit","NRAS proto-oncogene, GTPase",null,"leukotriene C4 synthase","ankyrin repeat domain 39","queuine tRNA-ribosyltransferase catalytic subunit 1","component of inhibitor of nuclear factor kappa B kinase complex","MAX dimerization protein 3","glutathione S-transferase mu 2","component of oligomeric golgi complex 8","Rho GTPase activating protein 19","HAUS augmin like complex subunit 7","lecithin-cholesterol acyltransferase","protein tyrosine phosphatase receptor type C associated protein","PVR related immunoglobulin domain containing","glypican 2","signal-induced proliferation-associated 1","endogenous retrovirus group 3 member 1, envelope","synaptojanin 2 binding protein","ADP ribosylation factor like GTPase 2","tubulin tyrosine ligase like 13, pseudogene","guanylate binding protein 7","RBMX like 1","steroid receptor RNA activator 1","STIM activating enhancer","DnaJ heat shock protein family (Hsp40) member C9","chromosome 8 open reading frame 82","voltage dependent anion channel 1","zinc finger and BTB domain containing 9","thioredoxin related transmembrane protein 2","hexosaminidase subunit alpha","NADH:ubiquinone oxidoreductase core subunit S3","leptin receptor overlapping transcript","LBH regulator of WNT signaling pathway","adenosine deaminase tRNA specific 3","protein phosphatase 1 catalytic subunit beta","sulfotransferase family 1A member 4","G protein signaling modulator 3","linker for activation of T cells","NCK interacting protein with SH3 domain","activating transcription factor 6 beta","three prime repair exonuclease 1","sphingosine-1-phosphate receptor 3","solute carrier family 35 member F6","chloride intracellular channel 1","dimethylarginine dimethylaminohydrolase 2","ribosomal protein S29","zinc finger protein 134","general transcription factor IIH subunit 4","DEAD-box helicase 47","zinc finger protein 888","zinc finger protein 845","killer cell lectin like receptor K1","epithelial membrane protein 2","potassium channel tetramerization domain containing 11","ubiquitin D","protein phosphatase, Mg2+/Mn2+ dependent 1N (putative)","solute carrier family 23 member 3","leukotriene B4 receptor","leukotriene B4 receptor 2","deoxyribonuclease 1","magnesium dependent phosphatase 1","casein kinase 1 epsilon","interferon regulatory factor 9","galactose-1-phosphate uridylyltransferase","claudin 9","integrin subunit alpha 1","nudix hydrolase 19","zinc finger protein 726","zinc finger protein 99","Tax1 binding protein 3","adaptor related protein complex 1 subunit gamma 2","zinc finger protein 90","NAD(P)HX dehydratase","BORCS8-MEF2B readthrough","glucosidase alpha, neutral C","tubulin tyrosine ligase like 3","replication initiator 1","mitochondrial ribosomal protein L23","zinc finger protein 891","small integral membrane protein 7","F-box protein 16","tetraspanin 4","copine 1","ADP ribosylation factor like GTPase 16","WEE2 oocyte meiosis inhibiting kinase","LYR motif containing 4","MYC binding protein","transmembrane protein 213","photoreceptor disc component","ALG3 alpha-1,3- mannosyltransferase","SH3 domain containing 21","small integral membrane protein 30","chromosome 19 open reading frame 38","chromosome 17 open reading frame 67","fission, mitochondrial 1",null,"angiogenin","scavenger receptor family member expressed on T cells 1","colorectal cancer associated 2","speedy/RINGO cell cycle regulator family member E3","metallo-beta-lactamase domain containing 1","SOGA family member 3","neuralized E3 ubiquitin protein ligase 1B","HAUS augmin like complex subunit 3","BBSome interacting protein 1","arsenite methyltransferase","coiled-coil domain containing 103","perilipin 5","SEC14 like lipid binding 6","notochord homeobox","protein phosphatase methylesterase 1","StAR related lipid transfer domain containing 10","NUT family member 2D","cytoplasmic polyadenylation element binding protein 1","EMAP like 6","zinc finger protein 727",null,"zinc finger SWIM-type containing 8","chromosome 10 open reading frame 105","Rho guanine nucleotide exchange factor 33","interferon related developmental regulator 2","calpain 14","zinc finger BED-type containing 1","translocase of outer mitochondrial membrane 6","heterogeneous nuclear ribonucleoprotein U like 2","citrate synthase lysine methyltransferase","membrane spanning 4-domains A4E","fer-1 like family member 6","mature T cell proliferation 1","doublecortin domain containing 2C","smoothelin like 1","nuclear pore complex interacting protein family member A8","zinc finger SWIM-type containing 7","Rho guanine nucleotide exchange factor 28","TBC1 domain family member 26","leucine rich repeat containing 69","CDP-L-ribitol pyrophosphorylase A","nuclear pore complex interacting protein family member A7","retrotransposon Gag like 10","collagen type XXVIII alpha 1 chain","prohibitin 2","t-complex 11 family, X-linked 2","neuralized E3 ubiquitin protein ligase 4","Grid2 interacting protein","UBX domain protein 2B","COBW domain containing 6","mucin 5AC, oligomeric mucus/gel-forming","family with sequence similarity 166 member B","peroxisomal biogenesis factor 26","chromosome 5 open reading frame 49","FAST kinase domains 5","golgin A8 family member B","homeobox and leucine zipper encoding","DEAD-box helicase 3 X-linked","VPS16 core subunit of CORVET and HOPS complexes","myosin light chain 5","zinc finger protein 407","aminopeptidase like 1","RPL17-C18orf32 readthrough","siah E3 ubiquitin protein ligase family member 3","EF-hand calcium binding domain 8","regulator of solute carriers 1","transmembrane protein 242","transmembrane protein 167B","family with sequence similarity 72 member D","TNF receptor superfamily member 25","solute carrier family 35 member E2A","thiosulfate sulfurtransferase like domain containing 1","repetin","cytochrome b5 reductase like","chromosome 1 open reading frame 167","tetratricopeptide repeat domain 34","ATPase family AAA domain containing 3C","IFI30 lysosomal thiol reductase","coiled-coil domain containing 7","folliculin interacting protein 1","chemokine like factor","presequence translocase associated motor 16","cell division cycle 14C","CEBPZ opposite strand","zinc finger protein 579","chymotrypsin like elastase 3B","ribonuclease K","NBPF member 1","UBAP1-MVB12-associated (UMA) domain containing 1","family with sequence similarity 228 member B","leucine rich repeat and Ig domain containing 3","zinc finger GATA like protein 1","vesicle associated membrane protein 2","olfactory receptor family 6 subfamily B member 1","chromosome 6 open reading frame 226","protein phosphatase 3 regulatory subunit B, alpha","FA complementation group G","olfactory receptor family 2 subfamily A member 5","adaptor related protein complex 4 subunit mu 1","chromosome 2 open reading frame 16","olfactory receptor family 2 subfamily A member 12","CCAAT enhancer binding protein delta","ZNF816-ZNF321P readthrough","ariadne RBR E3 ubiquitin protein ligase 2 opposite strand","zinc finger BED-type containing 8","olfactory receptor family 1 subfamily C member 1","neuronal pentraxin receptor","family with sequence similarity 200 member A","protein phosphatase 2 regulatory subunit Balpha","zinc finger protein 880","tripartite motif containing 16","tigger transposable element derived 1","XK related 9","solute carrier family 12 member 8","apolipoprotein L6","fatty acid desaturase 3","olfactory receptor family 2 subfamily A member 1","cyclin L2","ubiquitin A-52 residue ribosomal protein fusion product 1","palmitoyl-protein thioesterase 2","zinc finger protein 630","TGFB1-induced anti-apoptotic factor 1","BTB domain containing 19","family with sequence similarity 185 member A","POTE ankyrin domain family member M","doublecortin domain containing 2B","chromosome 10 open reading frame 55","exosome component 6","VPS52 subunit of GARP complex","zinc finger protein 844","creatine kinase, mitochondrial 1A","TINCR ubiquitin domain containing","growth differentiation factor 1","major histocompatibility complex, class II, DP beta 1","C1q and TNF related 5","ceramide-1-phosphate transfer protein","proline rich 29","complement C4B (Chido blood group)","ataxin 1 like","small integral membrane protein 13",null,"nuclear pore complex interacting protein family member A3","NADH:ubiquinone oxidoreductase complex assembly factor 8","transmembrane protein 233","pleckstrin homology and RUN domain containing M1","cancer/testis antigen 62","MKRN2 opposite strand","C2 calcium dependent domain containing 4D","zinc finger protein 469","MAPK regulated corepressor interacting protein 1","solute carrier family 26 member 6","family with sequence similarity 229 member A","ERCC excision repair 6, chromatin remodeling factor","nucleolar protein 7","CTAGE family member 4","PIGB opposite strand 1","formiminotransferase cyclodeaminase N-terminal like","testis expressed 22","olfactory receptor family 52 subfamily I member 2","ciliary rootlet coiled-coil, rootletin family member 2","transmembrane protein 185B","uncharacterized LOC102725191","heat shock factor binding protein 1 like 1","serine/arginine repetitive matrix 5","phosphoglycerate mutase family member 4","chromosome 13 open reading frame 42","endogenous retrovirus group MER34 member 1, envelope","chromosome 14 open reading frame 132","WD repeat domain 46","zinc finger protein 717","killin, p53 regulated DNA replication inhibitor","poly(ADP-ribose) glycohydrolase","aldo-keto reductase family 1 member B15","secretory carrier membrane protein 4","lymphotoxin beta","myoregulin","RNA polymerase II subunit J2","olfactory receptor family 2 subfamily M member 3","ATP synthase F0 subunit 8","orosomucoid 2","family with sequence similarity 174 member C","oligosaccharyltransferase complex subunit 4, non-catalytic","chromosome 2 open reading frame 92","NUT family member 2E","fibronectin type III domain containing 10","proline rich basic protein 1","ADP ribosylation factor like GTPase 17A","dihydrofolate reductase","suppressor APC domain containing 1","ribosomal protein L41","orosomucoid 1","PAT1 homolog 2","zinc finger protein 492","zinc finger protein 688","PET100 cytochrome c oxidase chaperone","glycerol kinase 3 pseudogene","family with sequence similarity 90 member A26","IQ motif containing F3","ankyrin repeat domain 66","acyl-CoA binding domain containing 6","nuclear factor, erythroid 4",null,"ankyrin repeat domain 18B","PPP5 tetratricopeptide repeat domain containing 1","coiled-coil domain containing 192","proline rich protein BstNI subfamily 4","YY2 transcription factor","stathmin domain containing 1","heat shock factor binding protein 1","olfactory receptor family 5 subfamily H member 1","CFAP97 domain containing 1","major histocompatibility complex, class II, DP alpha 1","ribosomal protein S18","tetraspanin 19","cytochrome P450 family 21 subfamily A member 2","proline rich protein HaeIII subfamily 1","TAP binding protein","zinc finger BED-type containing 9","translation machinery associated 7 homolog","testis expressed 50","MCTS1 re-initiation and release factor","MT-RNR2 like 4","olfactory receptor family 52 subfamily I member 1","small integral membrane protein 26","apical junction component 1 homolog",null,"major histocompatibility complex, class II, DQ beta 2","golgin A8 family member N","tumor necrosis factor","PET117 cytochrome c oxidase chaperone","LYR motif containing 9",null,"ring finger protein 224",null,"BTB domain containing 18","transmembrane protein 238",null,"H2B clustered histone 15","ribosomal protein S28","ubiquinol-cytochrome c reductase hinge protein like","tripartite motif containing 26","zinc finger protein 879","coiled-coil domain containing 188","zinc finger protein 736","phospholipase A2 inhibitor and LY6/PLAUR domain containing","chromosome 5 open reading frame 58","family with sequence similarity 133 member B","multiciliate differentiation and DNA synthesis associated cell cycle protein","Jrk helix-turn-helix protein","nuclear pore complex interacting protein family member B2","major histocompatibility complex, class I, B","IQ motif containing M","HNRNPUL2-BSCL2 readthrough (NMD candidate)","shisa family member 8","ankyrin repeat domain 65","BRD3 opposite strand","zinc finger and SCAN domain containing 31","chromosome 12 open reading frame 75","HGH1 homolog","protein phosphatase 1 regulatory subunit 3E",null,"small regulatory polypeptide of amino acid response","small integral membrane protein 27","NFAT activating protein with ITAM motif 1","ankyrin repeat domain 34C","KIAA0040","beta-1,3-galactosyltransferase 4","olfactory receptor family 5 subfamily H member 14","zinc finger and BTB domain containing 22","zinc finger BED-type containing 5","peptidylprolyl isomerase A like 4G","coiled-coil domain containing 200","ubiquitin conjugating enzyme E2 L5","zinc finger protein 853","Rho guanine nucleotide exchange factor 38","CTAGE family member 9","UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 9","CDKN2A interacting protein N-terminal like","creatine kinase, mitochondrial 1B","ring finger protein 223","prostate and testis expressed 4","zinc finger protein 737","ral guanine nucleotide dissociation stimulator like 2","chromosome 10 open reading frame 143","shisa family member 9","olfactory receptor family 7 subfamily E member 24","major histocompatibility complex, class II, DQ alpha 1","kinesin family member C1","chromosome 2 open reading frame 74","immunity related GTPase M","amylase alpha 1A","family with sequence similarity 200 member B",null,"leucine rich repeat containing 37 member A2","transmembrane protein 250","thioredoxin domain containing 5","cytosolic arginine sensor for mTORC1 subunit 1","ring finger protein 103","RNA binding motif protein 14","alkB homolog 6","ankyrin repeat and SOCS box containing 14","kelch like family member 41","CASTOR family member 3","sperm-tail PG-rich repeat containing 4","NME/NM23 nucleoside diphosphate kinase 1","TNF superfamily member 12","CMT1A duplicated region transcript 4","apolipoprotein B mRNA editing enzyme catalytic subunit 3G","toll like receptor 9","WW domain binding protein 1","mitochondrial ribosomal protein S17","guided entry of tail-anchored proteins factor 4","chromosome 1 open reading frame 226","adenylosuccinate lyase",null,"leukocyte immunoglobulin like receptor A2","testis expressed 35","amylase alpha 2B","small transmembrane regulator of ion transport 1","lymphocyte antigen 6 family member G5B","proteasome 20S subunit beta 9","protocadherin gamma subfamily C, 3","small lysine rich protein 1","UDP glucuronosyltransferase family 1 member A5","cytochrome c oxidase assembly factor COX19","acyl-CoA dehydrogenase family member 11","peptidylprolyl isomerase like 3","forkhead box O3B","TNF receptor superfamily member 13B","aquaporin 1 (Colton blood group)","C1q and TNF related 9","ISY1 splicing factor homolog","PNMA family member 2","leucine rich repeats and death domain containing 1","protocadherin gamma subfamily C, 5","transmembrane protein 189","retinol dehydrogenase 14","phosphatidylinositol specific phospholipase C X domain containing 2","macrophage migration inhibitory factor","NOP2/Sun RNA methyltransferase 6","major histocompatibility complex, class II, DO beta","YAE1 maturation factor of ABCE1","teratocarcinoma-derived growth factor 1","chromosome 3 open reading frame 85","CGRP receptor component","CMT1A duplicated region transcript 1","ribosomal protein L36a","pyridoxal phosphatase","ribonuclease P/MRP subunit p21","CD302 molecule","EGF like domain multiple 8","ATP synthase membrane subunit f","Rho GTPase activating protein 8",null,"actin related protein 2/3 complex subunit 4","cortistatin","UDP glucuronosyltransferase family 1 member A1","indolethylamine N-methyltransferase","actin related protein 2/3 complex subunit 1A","small proline rich protein 2A","ATP synthase peripheral stalk subunit OSCP","pleckstrin homology domain containing O2","chromosome 8 open reading frame 58","phosphatidylserine decarboxylase","4-hydroxy-2-oxoglutarate aldolase 1","PWP2 small subunit processome component",null,"phosphatidylinositol 4-kinase alpha","huntingtin interacting protein K","alpha-methylacyl-CoA racemase","mitochondrial fission process 1","t-complex 10 like","pregnancy specific beta-1-glycoprotein 2","ADP ribosylation factor GTPase activating protein 3","chromosome 22 open reading frame 39","UDP glucuronosyltransferase family 1 member A8","eukaryotic translation initiation factor 6","protocadherin gamma subfamily C, 4","mitochondrial ribosomal protein L20","actin related protein 2/3 complex inhibitor","UDP glucuronosyltransferase family 1 member A10","serpin family B member 10","major histocompatibility complex, class II, DM beta","2,4-dienoyl-CoA reductase 2","G protein subunit gamma 10","ciliary neurotrophic factor","coiled-coil domain containing 169","retrotransposon Gag like 5","adaptor related protein complex 5 subunit zeta 1","zinc finger protein 709","stereocilin","endogenous retrovirus group W member 1, envelope","eukaryotic translation initiation factor 4E binding protein 3","pregnancy specific beta-1-glycoprotein 11","UDP glucuronosyltransferase family 1 member A3","mitochondrial ribosomal protein L33","microtubule associated monooxygenase, calponin and LIM domain containing 3","PPAN-P2RY11 readthrough","protocadherin alpha subfamily C, 2","stonin 1","PRA1 domain family member 2","V-set and immunoglobulin domain containing 8","short transmembrane mitochondrial protein 1","potassium channel tetramerization domain containing 7","ephrin A4","toll like receptor adaptor molecule 2","N(alpha)-acetyltransferase 80, NatH catalytic subunit","TNF receptor superfamily member 6b","WAP four-disulfide core domain 6","uroplakin 3B","small integral membrane protein 34A","interleukin 10 receptor subunit beta","complement factor B","zinc finger protein 487","WD repeat domain 92","NME/NM23 nucleoside diphosphate kinase 2",null,"phospholipase A2 group IVB","cilia and flagella associated protein 57","nuclear pore complex interacting protein family member B5","tetratricopeptide repeat domain 4","transmembrane protein 35B","jumonji domain containing 7","apolipoprotein B mRNA editing enzyme catalytic subunit 3D","olfactory receptor family 2 subfamily A member 7","tubulin alpha 4b","mitochondrial ribosomal protein S6","zinc finger protein 512","glutathione S-transferase alpha 1","aminoacylase 1","NFS1 cysteine desulfurase","dolichyl-diphosphooligosaccharide--protein glycosyltransferase non-catalytic subunit","transmembrane protein 199","glutathione S-transferase alpha 2","DNAJC25-GNG10 readthrough","UDP glucuronosyltransferase family 1 member A7","purinergic receptor P2Y11","transmembrane protein 141","transmembrane protein 225B","interferon induced transmembrane protein 10","dysbindin domain containing 2","ETS variant transcription factor 5","RNA binding motif protein 12","UDP glucuronosyltransferase family 1 member A4","endogenous retrovirus group FRD member 1, envelope","leukocyte immunoglobulin like receptor A6","scavenger receptor class F member 2","apolipoprotein B mRNA editing enzyme catalytic subunit 3C","coiled-coil domain containing 13","aspartic peptidase retroviral like 1","ubiquitin conjugating enzyme E2 V1","CTAGE family member 8","patched domain containing 4","complement C4A (Rodgers blood group)","hemoglobin subunit beta","crystallin beta B2","NEDD4 binding protein 2 like 2","zinc finger protein 585B","CCAAT enhancer binding protein alpha","H2A.J histone","ubiquitin associated protein 1 like","PGAM family member 5, mitochondrial serine/threonine protein phosphatase","zinc finger CCHC-type containing 3","SPTY2D1 opposite strand","twinfilin actin binding protein 2","methionyl-tRNA synthetase 2, mitochondrial","ubiquitin specific peptidase 51","branched chain keto acid dehydrogenase E1 subunit alpha","insulin like 3","alcohol dehydrogenase 1C (class I), gamma polypeptide","apelin receptor early endogenous ligand","cyclin dependent kinase 11B","protocadherin alpha subfamily C, 1","POU domain class 5, transcription factor 2","Purkinje cell protein 4 like 1","abhydrolase domain containing 14A","STIMATE-MUSTN1 readthrough","RBM14-RBM4 readthrough","LY75-CD302 readthrough","TRIM59-IFT80 protein","coiled-coil domain containing 153","small integral membrane protein 31","TNFSF12-TNFSF13 readthrough","formin 1","HAUS augmin like complex subunit 5","EPPIN-WFDC6 readthrough",null,"ATP synthase membrane subunit g like",null,"transmembrane protein 150C",null,"NLR family apoptosis inhibitory protein","zinc finger protein 324B","spermatogenesis associated serine rich 1","interferon alpha and beta receptor subunit 2","zinc finger protein 564","fer-1 like family member 5",null,"RNF103-CHMP3 readthrough","programmed cell death 6","telomere repeat binding bouquet formation protein 1","transmembrane protein 158 (gene/pseudogene)","ARPIN-AP3S2 readthrough","YjeF N-terminal domain containing 3","protocadherin alpha 10","ARPC4-TTLL3 readthrough",null,"tRNA methyltransferase 9B (putative)","zinc finger protein 718","small integral membrane protein 20","TCL1 upstream neural differentiation-associated RNA","coiled-coil-helix-coiled-coil-helix domain containing 10","cyclin dependent kinase 3","G protein-coupled receptor 162","ATPase H+ transporting V1 subunit E2","GLI family zinc finger 4",null,"reeler domain containing 1","selenoprotein P","zinc finger protein 474-like","zinc finger protein 674","TMED7-TICAM2 readthrough","zinc finger protein 345","ALG1 chitobiosyldiphosphodolichol beta-mannosyltransferase like 2","SH3 and multiple ankyrin repeat domains 3","zinc finger protein 550","CENPS-CORT readthrough",null,null,"protocadherin gamma subfamily A, 12","shieldin complex subunit 3","coiled-coil domain containing 71 like","transmembrane protein 200B","protocadherin gamma subfamily B, 6","serpin family E member 3","chromosome 1 open reading frame 210","TMF1 regulated nuclear protein 1","small integral membrane protein 18","protocadherin gamma subfamily A, 5","protocadherin gamma subfamily A, 7","solute carrier family 10 member 5","eukaryotic translation initiation factor 5A like 1",null,"ALG11 alpha-1,2-mannosyltransferase","ataxin 7 like 3B","protein kinase, DNA-activated, catalytic subunit","protocadherin gamma subfamily A, 6","protocadherin gamma subfamily A, 8","UTP14C small subunit processome component","protocadherin gamma subfamily A, 10","protocadherin gamma subfamily A, 11","protocadherin gamma subfamily B, 2","protocadherin gamma subfamily B, 4","claudin 23","zinc finger protein 260","LYN proto-oncogene, Src family tyrosine kinase","PIN2 (TERF1) interacting telomerase inhibitor 1","protocadherin gamma subfamily B, 7","CD8b2 molecule","nuclear pore complex interacting protein family member B11","CHKB-CPT1B readthrough (NMD candidate)","sialic acid binding Ig like lectin 14","prostate and breast cancer overexpressed 1",null,"adaptor related protein complex 5 subunit beta 1","charged multivesicular body protein 4A",null,null,"fucose-1-phosphate guanylyltransferase",null,"immunoglobulin lambda like polypeptide 5","mex-3 RNA binding family member A",null,"olfactory receptor family 10 subfamily G member 4","eukaryotic translation elongation factor 1 gamma","SYS1-DBNDD2 readthrough (NMD candidate)","solute carrier family 22 member 18 antisense","nuclear pore complex interacting protein family member A2","MPV17 mitochondrial inner membrane protein like 2","ATP6V1G2-DDX39B readthrough (NMD candidate)","BLOC-1 related complex subunit 8","INMT-MINDY4 readthrough (NMD candidate)","dipeptidyl peptidase 3","STX16-NPEPL1 readthrough (NMD candidate)","ANKHD1-EIF4EBP3 readthrough","BRICK1 subunit of SCAR/WAVE actin nucleating complex","SAA2-SAA4 readthrough",null,"charged multivesicular body protein 1B","EP300 interacting inhibitor of differentiation 3","MSH5-SAPCD1 readthrough (NMD candidate)","hydroxyacyl-thioester dehydratase type 2","Nanog homeobox retrogene P8","caspase recruitment domain family member 17",null,"EP300 interacting inhibitor of differentiation 1",null,"NADPH oxidase 5","hydroxycarboxylic acid receptor 3","endogenous Bornavirus like nucleoprotein 2","terminal uridylyl transferase 1, U6 snRNA-specific","RNA polymerase II subunit M","ferredoxin-fold anticodon binding domain containing 1","RAB44, member RAS oncogene family",null,"olfactory receptor family 6 subfamily J member 1 (gene/pseudogene)",null,"MT-RNR2 like 8","TIFA inhibitor","taste 2 receptor member 20",null,"cytochrome P450 family 2 subfamily A member 6","small nucleolar RNA host gene 28","cathepsin O","cytochrome c oxidase assembly factor 8","trafficking protein particle complex 2B","dynein axonemal assembly factor 4","zinc finger protein 432","taste 2 receptor member 30","zinc finger protein 10","zinc finger protein 486","small integral membrane protein 3","hydroxymethylbilane synthase","zinc finger protein 225",null,"taste 2 receptor member 31","DND microRNA-mediated repression inhibitor 1","spalt like transcription factor 3",null,"DNA polymerase gamma 2, accessory subunit","small integral membrane protein 10 like 1",null,null,"LIM zinc finger domain containing 4","zinc finger protein 350","zinc finger protein 253","chromosome 17 open reading frame 100","solute carrier family 5 member 8","LIM zinc finger domain containing 3","haptoglobin","chromosome 11 open reading frame 97","DENN domain containing 11","LSM14A mRNA processing body assembly factor","NHL repeat containing 4","glutamyl-tRNA amidotransferase subunit C","zinc finger BED-type containing 6","maltase-glucoamylase",null,"farnesyltransferase, CAAX box, beta",null,"ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 6","zinc finger protein 625","polypeptide N-acetylgalactosaminyltransferase 4","InaF motif containing 1","canopy FGF signaling regulator 2","maltase-glucoamylase 2 (putative)",null,"cut like homeobox 1","TEN1 subunit of CST complex","P2RX5-TAX1BP3 readthrough (NMD candidate)","microtubule associated protein 1 light chain 3 beta 2","C-type lectin domain containing 5A","churchill domain containing 1",null,"chromosome 17 open reading frame 49","regulator of telomere elongation helicase 1","PPT2-EGFL8 readthrough (NMD candidate)","zinc finger protein 578","peptide deformylase, mitochondrial",null,null,null,"sperm equatorial segment protein 1",null,"SPECC1L-ADORA2A readthrough (NMD candidate)","BCL2L2-PABPN1 readthrough","SYNJ2BP-COX16 readthrough",null,"tripartite motif containing 34","protein only RNase P catalytic subunit","ribonuclease A family member 4","melanocortin 1 receptor",null,"centrosomal protein 95","tubulin beta 3 class III","UBE2F-SCLY readthrough (NMD candidate)",null,"TVP23C-CDRT4 readthrough","FPGT-TNNI3K readthrough","BLOC1S5-TXNDC5 readthrough (NMD candidate)","POC1B-GALNT4 readthrough","NDUFC2-KCTD14 readthrough","small integral membrane protein 6",null,"integrin subunit beta 3","solute carrier family 35 member G6","ZHX1-C8orf76 readthrough",null,"InaF motif containing 2","ST20-MTHFS readthrough","cortexin domain containing 1","thiamine triphosphatase","mitochondrial ribosomal protein L46","ubiquitin conjugating enzyme E2 Q2 like",null,null,"RNA binding motif protein 15B","transforming growth factor beta receptor 3 like",null,"speedy/RINGO cell cycle regulator family member E6","coiled-coil domain containing 187","ferric chelate reductase 1 like","PMF1-BGLAP readthrough","mannose receptor C-type 1","scleraxis bHLH transcription factor","chromosome 16 open reading frame 95",null,"double homeobox 4",null,"ankyrin repeat domain 20 family member A1",null,null,"cell cycle progression 1","sulfotransferase family 1A member 3","transmembrane protein 178B","MPV17 mitochondrial inner membrane protein like",null,"epiplakin 1","C-type lectin domain containing 19A","zinc finger protein 865","BOP1 ribosomal biogenesis factor","mucin 22","uncharacterized LOC105372440","platelet and endothelial cell adhesion molecule 1","TEN1-CDK3 readthrough (NMD candidate)","tubulin beta 8 class VIII",null,"gigaxonin",null,"golgin A6 family like 7","chromosome 15 open reading frame 65",null,"golgin A8 family member S","BOLA2-SMG1P6 readthrough","transcription factor 24","golgin A8 family member H","ISY1-RAB43 readthrough","CLN3 lysosomal/endosomal transmembrane protein, battenin","MIA SH3 domain containing",null,null,"protocadherin gamma subfamily A, 9",null,"protocadherin gamma subfamily B, 3","coronin 7",null,null,"matrix metallopeptidase 12","speedy/RINGO cell cycle regulator family member E17",null,"spondin 1","OVCA2 serine hydrolase domain containing","mitochondrial ribosomal protein L12","cyclin Q","general transcription factor IIi","zinc finger protein 234",null,"myocardial zonula adherens protein",null,"transmembrane protein 238 like","serine and arginine rich splicing factor 8","inhibitor of nuclear factor kappa B kinase subunit epsilon",null,"microseminoprotein beta","corticotropin releasing hormone receptor 1","long intergenic non-protein coding RNA 672","NBPF member 11","regulator of hemoglobinization and erythroid cell expansion","aldo-keto reductase family 1 member C8, pseudogene",null,"annexin A8 like 1",null,"notch 2 N-terminal like A","dynein light chain LC8-type 2","OTU deubiquitinase 7B",null,null,"annexin A8","RNA binding motif protein 8A","translocase of inner mitochondrial membrane 23","ring finger protein 115","C21orf59-TCP10L readthrough","ribosomal protein L17",null,"SEC22 homolog B, vesicle trafficking protein (gene/pseudogene)","RAD54 homolog B","thioredoxin interacting protein","SLIT-ROBO Rho GTPase activating protein 2","BAH domain and coiled-coil containing 1",null,"Ras association domain family member 5","STE20 related adaptor alpha",null,"NBPF member 15","nuclear receptor coactivator 4","mitochondrial ribosomal protein S21","myosin XVB","FXYD domain containing ion transport regulator 1","alanyl-tRNA synthetase domain containing 1","zinc finger protein 850","prostaglandin E synthase 3 like",null,null,"chromosome 17 open reading frame 113","neuropeptide FF-amide peptide precursor",null,"uroplakin 3B like 1",null,null,"zinc finger protein 285","sphingosine-1-phosphate receptor 2","RNA polymerase II subunit J2","ferredoxin 2","zinc finger protein 224",null,"epididymal protein 13",null,"small integral membrane protein 22","lin-37 DREAM MuvB core complex component","NADH:ubiquinone oxidoreductase subunit A7","NBPF member 12","solute carrier family 6 member 14",null,"small integral membrane protein 17","t-complex 11 family, X-linked 1",null,"family with sequence similarity 156 member A",null,null,null,null,null,"Rho/Rac guanine nucleotide exchange factor 18","endogenous retrovirus group V member 2, envelope","MT-RNR2 like 2","F-box protein 17","thymosin beta 15B",null,"MAGI family member, X-linked","inhibitor of nuclear factor kappa B kinase regulatory subunit gamma","zinc finger protein 587B","Spi-B transcription factor","nuclear RNA export factor 2","nuclear RNA export factor 2B","transmembrane protein 185A",null,null,"NBPF member 9","coiled-coil domain containing 194","solute carrier family 25 member 53",null,"egl-9 family hypoxia inducible factor 2","COMMD3-BMI1 readthrough","FMC1-LUC7L2 readthrough","ZNF559-ZNF177 readthrough","TSNAX-DISC1 readthrough (NMD candidate)",null,"NCBP2 antisense 2 (head to head)","H4 clustered histone 15",null,null,"HEAT repeat containing 9","NBPF member 14","TATA-box binding protein associated factor 15","growth arrest specific 2 like 2","RPS10-NUDT3 readthrough","H4 clustered histone 14","RAS like family 10 member B","TLCD4-RWDD3 readthrough","UDP glucuronosyltransferase family 2 member A2","sulfiredoxin 1","NBPF member 19","NBPF member 10","matrix metallopeptidase 28","C-C motif chemokine ligand 5","peptidylprolyl isomerase A like 4E","limb and CNS expressed 1 like","mast cell immunoglobulin like receptor 1","MROH7-TTC4 readthrough (NMD candidate)",null,"ankyrin repeat domain 34A","general transcription factor IIH subunit 5","H2A clustered histone 19",null,"nudix hydrolase 3","lysine methyltransferase 2B","POM121 transmembrane nucleoporin C","CD24 molecule",null,"FAM47E-STBD1 readthrough",null,"cilia and flagella associated protein 206","zinc finger protein 595","double C2 domain beta","protocadherin beta 16","decapping mRNA 1A","ATP6V1F neighbor",null,null,"chromosome 2 open reading frame 15","glutamate ionotropic receptor NMDA type subunit 2B","NBPF member 26",null,"spermatogenesis associated 13","SNRPN upstream reading frame","H3.2 histone (putative)","zinc finger and BTB domain containing 8B","C1QTNF3-AMACR readthrough (NMD candidate)",null,"speedy/RINGO cell cycle regulator family member E17","ATP/GTP binding protein like 1","CWC25 spliceosome associated protein homolog","small integral membrane protein 11A","elongin BC and polycomb repressive complex 2 associated protein","zinc finger HIT-type containing 3","H2B clustered histone 14","cytoplasmic FMR1 interacting protein 1","H2B clustered histone 8","ubiquitin specific peptidase 27 X-linked","TATA-box binding protein associated factor 9","nucleolar protein 12","golgin A6 family like 1","H3 clustered histone 8","cytosolic arginine sensor for mTORC1 subunit 2","N-acetyltransferase domain containing 1","suppressor of cytokine signaling 7","cystathionine beta-synthase like","adrenoceptor alpha 2B","H2B clustered histone 6",null,"zinc finger protein 658","transmembrane protein 269","TBC1 domain family member 3","RCC1 like","RIMS binding protein 3B","H4 clustered histone 6","H2B clustered histone 17","elongin A3","H3 clustered histone 6","TBC1 domain family member 3B","NPHP3-ACAD11 readthrough (NMD candidate)","proapoptotic nucleolar protein 1","TBC1 domain family member 3I",null,"H2A clustered histone 12","zinc finger protein 280B","MLLT6, PHD finger containing","protein phosphatase 4 regulatory subunit 3B","synergin gamma","nudix hydrolase 18","zinc finger protein 2","H4 clustered histone 13","C-C motif chemokine ligand 16","leukocyte receptor cluster member 9","C-C motif chemokine ligand 4","PEAK1 related, kinase-activating pseudokinase 1","H3 clustered histone 11","C-C motif chemokine ligand 18","Fc fragment of IgG binding protein","HNF1 homeobox B","periodic tryptophan protein 2 homolog",null,"XK related 5",null,null,"apoptosis antagonizing transcription factor","H2B clustered histone 9","H3 clustered histone 1","C-C motif chemokine ligand 15","RNA binding motif protein 27","PRH1-PRR4 readthrough","RIMS binding protein 3","Rho GTPase activating protein 23","tubulin gamma complex associated protein 5","U2 small nuclear RNA auxiliary factor 1 like 5","salt inducible kinase 1B (putative)","dual specificity phosphatase 14","ubiquitin like with PHD and ring finger domains 1","ORAI calcium release-activated calcium modulator 1","C-C motif chemokine ligand 4 like 2","C-C motif chemokine ligand 3 like 1",null,"H4 clustered histone 9","ankyrin repeat domain 20 family member A3","phosphoinositide-3-kinase regulatory subunit 6","transcriptional adaptor 2A","olfactory receptor family 4 subfamily E member 1 (gene/pseudogene)","potassium voltage-gated channel subfamily E regulatory subunit 1B","phosphatidylinositol-5-phosphate 4-kinase type 2 beta","H2A clustered histone 14","C-C motif chemokine ligand 14","H2B clustered histone 3","protocadherin gamma subfamily B, 5","RAB7B, member RAS oncogene family","dachshund family transcription factor 1","H2A clustered histone 16","glutathione S-transferase theta 4","H4 clustered histone 5","H2A clustered histone 8","ICOS ligand","tRNA-yW synthesizing protein 1 homolog B","coagulation factor VIII associated 3","H4 clustered histone 4","phosphatidylinositol glycan anchor biosynthesis class W","coagulation factor VIII associated 1","H2B clustered histone 7","polycomb group ring finger 2","family with sequence similarity 243 member B","SRC kinase signaling inhibitor 1","G protein-coupled receptor 179","myristoylated alanine rich protein kinase C substrate","zinc finger protein 670","polycystin 1 like 3, transient receptor potential channel interacting","glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1",null,"C-C motif chemokine ligand 3","p53-regulated lncRNA 1",null,"synaptotagmin-15","H3 clustered histone 7","proteasome 20S subunit beta 3","steroid 5 alpha-reductase 2",null,"CDGSH iron sulfur domain 3","RAD52 motif containing 1","DExD-box helicase 52","zinc finger protein 8","LOC110117498-PIK3R3 readthrough","somatostatin receptor 3","prickle planar cell polarity protein 4","myosin XIX","gametogenetin binding protein 2","zinc finger protein 229","H2A clustered histone 4","dehydrogenase/reductase 11","acetyl-CoA carboxylase alpha","transmembrane protein 191B","nuclear receptor subfamily 2 group E member 3","H2B clustered histone 10","chromosome 11 open reading frame 98","mitochondrial rRNA methyltransferase 1","H4 clustered histone 1","golgin A6 family like 10","elongin A3 family member B","H2A clustered histone 17","H4 clustered histone 2","H3 clustered histone 10","mitochondrial ribosomal protein L45",null,"olfactory receptor family 10 subfamily A member 6 (gene/pseudogene)","thiosulfate sulfurtransferase like domain containing 3","olfactory receptor family 2 subfamily T member 11 (gene/pseudogene)","olfactory receptor family 2 subfamily AG member 1 (gene/pseudogene)",null,"peptidylprolyl isomerase A like 4F","GVQW motif containing 2","glutamine amidotransferase like class 1 domain containing 3B","protocadherin 20",null,null,null,"coiled-coil domain containing 163","PAXIP1 associated glutamate rich protein 1","matrin 3",null,"transmembrane protein 272",null,null,null,"transmembrane protein 265",null,null,null,null,"schwannomin interacting protein 1",null,"chromosome 13 open reading frame 46","zinc finger protein 724",null,null,null,"small integral membrane protein 33","testis expressed 52",null,"CFAP97 domain containing 2","contactin associated protein like 3C","family with sequence similarity 240 member A","family with sequence similarity 95 member C","LITAF domain containing","proline rich transmembrane protein 1B","zinc finger CW-type and PWWP domain containing 2","chromosome 1 open reading frame 50","family with sequence similarity 169 member B","exocyst complex component 3 like 2","myocilin opposite strand","V-set and immunoglobulin domain containing 10 like 2",null,"presenilin associated rhomboid like",null,null,null,null,"TPTEP2-CSNK1E readthrough",null,"secreted LY6/PLAUR domain containing 2","heat shock protein family A (Hsp70) member 14","SCO cytochrome c oxidase assembly protein 2",null,"chromosome 2 open reading frame 81","LIMS3-LOC440895 readthrough",null,null,null,"LYNX1-SLURP2 readthrough",null,null,null,null,null,null,null,"EEF1A lysine methyltransferase 4","phosphodiesterase 8B","tubulin folding cofactor E",null,null,"small integral membrane protein 41","uncharacterized LOC729966",null,null,"coiled-coil domain containing 39","SEPT5-GP1BB readthrough","Rho GTPase activating protein 11B",null,null,null,null,null,"uroplakin 3B like 2",null,"uncharaterized LOC112694756","tubulin folding cofactor E",null,null,null,null,"chimerin 2",null,null,null,null,"zinc finger protein 280D","ataxin 7","solute carrier family 35 member D2","ATP binding cassette subfamily F member 2","COMM domain containing 7","RNA polymerase II subunit J3","superoxide dismutase 2",null,"zinc finger protein 883","chromosome 4 open reading frame 36",null,"zinc finger protein 365",null,null,null,null,null,null,null,null,"DNA polymerase alpha 2, accessory subunit",null,null,null,null,"ring finger protein 43",null,null,null,null,"leucine rich repeat containing 57","spindle and centriole associated protein 1","KRIT1 ankyrin repeat containing",null,null,"retinoic acid early transcript 1E",null,"notch 2 N-terminal like B",null,"gamma-glutamyltransferase 1",null,"zinc finger DHHC-type containing 11B","zinc finger protein 844","notch 2 N-terminal like R","kynurenine aminotransferase 1",null,"chromosome transmission fidelity factor 8","aryl-hydrocarbon receptor repressor",null,"uncharacterized LOC728392",null,"notch 2 N-terminal like C","speedy/RINGO cell cycle regulator family member E11",null,"zinc finger protein 761","FKBP prolyl isomerase 2","H3 clustered histone 2",null,"H3 clustered histone 3","HECT and RLD domain containing E3 ubiquitin protein ligase 3","two pore segment channel 2",null,null,null,null,null],[-0.7117278742511791,-0.4193350515024096,0.1788967056027135,-0.1768044924742686,1.174374871103207,0.5752875371242097,-0.214154605141823,-0.7421087512762889,0.250572539971526,-0.7079348478447054,-0.2098162388669138,-0.7455657300405892,-0.747226083470693,-0.4500561769454663,0.03028424233573103,0.01467317325560642,-0.484280813569233,-0.3775096105127554,-0.2089329246681921,-0.5747706846716782,1.854671437843759,-0.06438937276480246,0.09752593093589658,-0.4600317141061143,-0.3596640196614457,0.0510783903399681,0.3373783969155275,0.8669180196448968,1.199153100327791,0.5213760040390524,-1.148833812757078,-0.2497983101893105,-0.3836692105593063,-0.460546534766918,0.9027620954033091,0.8399295907544498,-0.05740379233087945,-0.01385638621364497,-0.04642207928654225,-0.6249702282769476,-1.41677767544387,-0.1843548346936315,-0.5541114871341536,-0.1555561311125696,1.180503913195632,0.05639761689642104,0.9995719759801907,0.354659578644115,-0.347422451499977,-0.141404218786374,0.002147819938237394,0.230224421267371,0.06689616268242392,-0.5650127135181038,-0.6239622601540561,-0.9605609971772058,-1.113051833259568,-0.8911289709400102,-0.453203809812137,-2.264074935908005,0.5416564010928867,-0.5253436513291048,0.2447375134142945,0.375414760013557,-0.2598425131824603,0.2924086301076674,0.1224788283225007,0.04861769382224394,0.3894742188991716,-0.3486711177052267,1.020892597770495,-0.2984911886987416,-0.1989601911526847,-0.4042833029100339,-0.2352918516889156,-0.3537284198179537,-0.2165774528154416,0.2709659524384623,-0.01030271909658863,-0.1651469151031574,0.191470131181593,-0.8609644659663239,-0.5003738240160521,0.2354239336569373,-0.07342764997319795,0.202416022377222,2.217568990849322,-0.3199409875230349,-0.913317750737317,-0.4901864334225565,-1.014905627390362,0.1471057799560875,-0.5281423001092117,-0.3062772047697226,-0.07022972557766904,0.09279262862516138,-0.3176095256327441,0.9276171433860435,-0.4598630676278014,-0.4073186762266745,0.07717462849091543,0.3502382384987378,-0.2009076463597199,0.03989643733960953,-0.3921183782276547,-0.1305776429557551,0.3363073920165235,0.1917938912921739,-0.6489122508687049,0.2294584871197237,0.1130774760980847,0.2648313266979394,0.5264256543318815,-0.1126680769621993,-0.2430015620968385,0.5949689396362183,-0.2811249068267374,-0.09788559072241908,0.1525855327814133,0.6007191451364846,-0.22528399689605,0.4537088838424947,-0.3152361174152587,-0.1961098468449582,0.2968777424076963,-0.2124981205873621,-0.8291456798200552,0.005418330458272522,-0.04810546733004251,-0.2936595067896611,-1.302450671307179,-0.4500534424534854,-0.02298023558096046,0.1628870682232617,-0.08286583991369305,-0.2383399316679785,-0.1030472965179853,-0.1180349021900243,0.222381949375754,-0.1585316606363364,-0.5299938173577232,-0.1295944669692924,-0.1450931720068044,-0.7599747147450295,-0.7915389962773109,0.5884292703636713,0.1658114027577062,-0.6875713947574401,0.1532091957022043,-0.9029971319014815,-0.2207364882850531,0.9570185322886395,0.1562159206179011,-0.1055829286898177,-0.8790475756596701,-0.1173444984342746,-0.5857894974186904,0.478361980913009,-0.6896123298911625,0.3783710580473155,1.118574542829008,-0.231142791036248,0.1934360141288089,-0.534278214014839,0.1170029665226327,0.1125582486236558,-0.2348467897253012,-0.005425749480681703,-0.804904835060138,-0.2588058726257739,-0.3955308081270938,-0.3060940485643354,0.03978047380155837,-0.007513999587821188,-0.7937318998331158,0.388489119979799,0.5170314325453843,1.446305608644821,-0.1516074866561337,0.1881816089345421,0.2106631548553835,0.3925705454352985,0.1302512987476188,1.129149126823472,-0.9525835756499248,0.04534573237138995,-0.5495690613723664,-0.6668828929933787,-0.4627103657041739,0.2525932807947537,0.2975880087222427,-0.549816417471907,0.181972340946658,-0.2778444405143105,-0.2606545506357505,-0.4753987502254285,-0.190087890370982,0.1439601872040976,-0.8089768024250159,0.6775662203780933,0.6851485542029024,1.496612439768267,-0.5959955789423278,-0.2462495004039696,-0.268418764247166,-0.1715940231665142,0.0496745705730005,-0.2157999725402679,-0.4319344467441824,-0.1578728374441833,-0.1027177667576587,-0.1540393596187017,0.7021226139357396,-0.7872646538095514,0.2046346677562057,-0.5787520514087421,0.09698438831603724,0.3023966835836509,-0.9200862571976365,-0.04601759777528088,-0.01021211395123234,-0.07167069784728204,0.04822641867122171,2.078385913300203,0.1387454275397214,-0.4154319818397793,-0.3975071971653529,0.1696616310868182,-0.304656409159664,0.3073191814762239,-0.6714591703921112,-0.5828340318098079,-0.09621147013695838,0.8215145646174568,-0.6330048846230403,-0.6653545065119991,-0.2891674028411823,1.554217950351368,-0.6894688250341724,-0.7729566337640477,-0.3991714256589804,0.599980489170585,0.814380843297744,-1.124739383154686,0.7670069836818133,0.2706460637799215,-0.2493050939562015,1.364024310504345,0.8337724571608738,-0.7550665725982753,-0.2480619014344539,-0.3299622244126333,-0.08061934389061615,-0.3447694942925856,-0.4953830778027158,-0.3363648403422367,0.2774666412434144,-0.5444952237539932,0.02455304037141202,-1.477464685570959,-0.2593511176400871,-0.2185652900014741,0.7373774129633085,-0.306986391913506,-0.3071284921508151,-0.02671663764420119,0.1416751574371539,0.01519126078041364,-0.2434116512228702,-0.3470408571387735,0.4885331192065513,0.1868400975413519,-0.07192462617397925,-0.3545838430290128,-0.1051133952996003,-0.05689894329455715,-0.5421604880713681,0.5811803228060362,-0.7897643853830086,0.08207456897200562,0.5981413757750847,-0.04412282560461506,-0.04228271005032177,-0.5116177239241562,1.204494639078298,0.3697486678283973,-0.174436798026468,-0.02650249609752878,-0.08623383703275737,-0.269174549767764,0.1572730342410553,0.96324886946073,-0.1563981790780162,-0.2893157253545652,0.007183076260563347,0.1414999894543116,0.1273249265773194,0.02857850979166414,-0.0617931366252804,0.1280415448599084,0.4577848789389163,-0.1299055445952879,0.346851078854799,1.247507875022949,-0.2894324554656045,-0.515919292116179,0.2160056092468697,-0.2264582268982017,-0.3086582209819813,-0.3398043849316493,0.3175664089608642,-0.1993966201806001,-0.2405754575959111,0.3953895362266547,-0.5861478663907046,0.2355353225439914,0.02675268416719359,-0.7412246028972554,0.2255461081913992,0.4542128895799755,-0.1389372302186346,0.5670114627775333,-0.4079660799886123,0.1233836800927601,-0.2833390323333154,-0.2798208981339923,0.0505862653312783,-0.5926064091714915,0.3911899549470282,0.3971581867340024,-0.2190355488633635,-0.3336149576802315,-0.6573642837446724,-0.3582942577970816,-0.2926396514161559,-0.2878693919192354,-0.2380393165800075,-0.6995418813750549,0.3716778250687147,-0.5336409545983311,0.03865155757104249,-0.1512123870700488,1.59724419823922,-0.2440701749581559,-0.4093876630292771,-1.641185058099578,0.1586937961784182,-0.661776870769621,-0.1377281722070545,-0.3916590742932896,-0.2574347151592699,-0.8063705889666023,-0.3514181899008202,0.08221308575818859,0.3525660220208823,-0.2459546079228747,0.2180845397225024,-1.661467506881965,0.7297799923245442,-0.1833451679253083,0.0223445235232093,-0.2740394158960426,-0.04144105047803022,-0.1096761859125132,-0.5896169515765537,0.6187986911460421,1.559777649447977,-0.3385811521899806,-1.171259195529668,0.7864064919646047,0.2870531047050534,-0.2753348813256727,0.07055411873826577,-0.5857147692429581,-0.190530146031502,-0.2393447998231755,0.2930495455076374,-0.4195591979448755,0.3119212126910205,-0.1300207515373167,0.7497633511344952,0.07029250365412743,0.08354430530020898,-0.3384921964403561,-0.1464430877010628,0.6872907935822373,-0.01707884368133395,-0.2303011539800645,-0.3023613311148917,0.3534120280418491,0.04374210109458729,1.27509296643595,-0.4160810974028891,-0.3545923938265568,0.2142163743862032,-0.3784941584491192,-0.4020606338916797,-0.1236230030679996,0.4094981599239911,1.290314866360872,-0.5340346354536352,0.01286873346865882,-0.01550930621121459,0.1273217159984595,-0.5464809107065115,0.0325222407220403,-0.7613811298212119,-0.09066302684275575,0.7640160828339284,1.078707404417527,0.4111021153993618,-0.6453565234389569,-0.239768142601854,-0.1698499524269785,1.126736485673764,0.4630548769464098,0.9069399799130733,0.2986182884639837,0.5963708505943652,0.2721801806402049,-0.1683123953314986,2.996464096907121,1.024672029319548,-1.19279657109071,0.6675403425823954,0.008672616565188303,-0.2677589340476109,1.775003393040629,0.1285825049712857,0.3524429727338686,-0.09301110822143649,0.3473356327674098,-0.2190634507984891,0.2169892205332033,0.9418642239713322,0.1639711099889321,-0.2204342571328288,-0.1756292376934835,0.001953095152203391,-0.2647655136680603,-0.06865376989524402,-0.5895149358356244,0.04528757445568093,0.5998296567793192,-0.3752195780557512,-0.3431885949331402,0.7359172478225359,-0.1069538446434135,1.271716074552665,-0.08856044995586503,-0.1663650342899132,0.0271227049858153,0.08819178375706692,-0.2099098093872296,0.2873951350157683,-0.9898220345781398,-0.4475633226616572,-0.02866539078596901,0.2027926640298551,0.06688164289126337,-0.08750007846620887,-0.1558575820789688,0.3498910471958248,0.3842394338397624,0.2158611169459885,0.1993585028881704,-0.2480637042239028,0.9550002312440234,0.1998821273660831,-1.197745902375554,0.7342256933239711,0.08309599410919201,0.2747049829824058,-0.5113444808276305,-0.07335002355567108,-0.6444049147877227,-0.225271133694716,0.1217535219607035,0.09198739660124916,-0.2882739403476977,0.4668375321026347,-0.5268465040555141,0.1660367628637786,0.6910724615483537,0.1402281722548731,-0.01312243364812245,0.09977628685123552,-0.401106497373309,-0.3241377310090912,0.08594651832896891,-0.3876025676672601,-0.9446436454548101,-0.1388866598327774,-0.3395036571816125,0.02624927966119725,0.103623793990686,-0.06294259156448888,0.09093515086429096,0.1213717294211646,-0.03435477429130532,0.788501892676304,-0.6346386185451968,0.5176213178611155,-0.1973152952200714,-0.1002184876067793,-1.15791423950427,0.1540335928369682,-0.03604787162194187,1.141209380297761,-0.0182743065676289,0.7440017125244804,-0.5161881539593132,-0.1406539077516213,-1.258634549307362,0.4796693182749694,0.2557721299461951,-0.6559075899531627,0.08033790760469926,-0.1597533991551794,-0.2697470564319897,-0.008353829761963583,-0.1295488612207103,0.08381142908622535,0.1835787469168998,0.418902206180471,0.3837512624691762,0.2662602887469437,0.1581763493772861,-0.5451102394872146,-0.27213628351434,-0.09009489094739903,-0.7783042749767719,0.1013331073373575,0.6678521322847337,-0.2599092937682477,-0.1162424936226031,0.03176040255089795,-0.6716704284291207,0.03461765396421496,-0.2046882329420011,0.8553411892037195,-0.05225676919197313,-1.557677986496623,-0.01330077097684235,-0.2800325096186072,-0.1732484321866148,-0.2528811131888559,0.3136588307452954,1.150337102535235,-0.9081471666799589,-0.6405345578362472,-0.3290162287917564,-0.1653711323236351,-0.1084944608500311,0.03597149869310425,-0.3215797495170744,0.3054118520177753,-0.5367605692929132,-0.7337181108373922,-0.2934816514960983,-1.102729968184456,-0.5725265376443948,0.1127321736465862,0.04617532182272576,-0.6217088123405174,0.2957285446691354,-0.2031618233388672,-0.3516172044752888,0.6748401570227597,-0.1164817211590082,-0.03147773498731934,-0.5048961669096073,0.5006003117121872,-0.7307224268078193,-0.1181987156383315,0.06582561770214522,-0.2596175756808143,0.08842558966576,-0.3204734358726957,0.7336047641619416,-0.2564847555486185,-0.3943380054602959,-0.1167839542423394,0.3694726490130402,-0.02836937824699231,-0.2418031170116887,-0.02001541991284838,0.2162321506489838,0.06419597298632465,0.06233021379371933,1.282389402813732,-0.2940179652552607,-0.1943087742262764,-0.1680444756209271,0.5788966974592935,-0.06063188749188106,0.1000194038181255,-0.2260398717652482,-0.4145281743665654,-0.5489872361191446,-0.3407589078762032,-0.04623608914669585,-0.04121919945141732,-0.5931317065711542,0.5621058443498046,-0.2163841037328476,0.3245377863161595,0.09611389628282445,-0.9203462033775469,0.2243069296159435,0.005567054780296162,-0.2375079034567878,-0.4198352954933673,0.6166148096816381,-0.5939286483889595,0.07999659087543495,-0.4602702382988949,-1.234656116290642,-0.250258296758924,-0.44189378147348,-0.3018271647320884,0.1980911779520783,-0.1698014067986806,-0.3538909307778764,-0.2752575215961598,-1.096707585024667,-0.3321354519952673,2.748605092324587,-0.430608814053475,0.2040376251949149,-0.598020037256483,-0.2026877522113434,0.2018153695628289,0.3405231891255282,-0.2092829006873784,-0.158387435785778,0.01515522935292754,0.4790194782434602,-0.4729650418721523,-0.1297531166936835,-0.3678225078559084,-1.091296462802525,0.02956266099614659,0.2663421772827958,-0.1985916717108522,1.279121245052134,-0.4008253206840003,-0.0246906566101466,-0.147184866358011,-0.4221444909737408,-0.4409506525060169,-0.2510827154100834,-0.3556105818342631,0.01895724720755003,0.1161571172417091,-0.5607610521678413,-0.5180512403240849,-0.1454277445841557,-0.4787470113313789,-0.1381749373478455,0.08299649078933688,-0.1141986342502295,-0.6172293865139792,-0.7401121672996402,-0.6112355397387724,0.7041369441238988,0.2518734207633906,-0.2785225636748172,-0.8555201046024677,0.001050668052113294,-0.1105580312013961,0.0803299315697151,-0.09702411801089703,-0.2871891519036368,-0.8588737059099931,0.06295226026742849,0.6544067728927845,0.2891695927352008,-0.02623410195506774,0.2269996482553351,0.1539197428123572,-0.001355145976461203,0.01136219109447811,0.2525431067806198,-0.5275097994835428,1.466469347326484,0.02277897374203087,0.2188143027617019,-0.127570905942753,-0.3738991518714331,-0.3548543805399734,0.3685716027400616,0.4345790579835673,-0.5355290012401533,-0.1307882641784961,-0.266170358975712,1.976188315277338,-0.03362315614396126,-0.4174329278394479,-0.259701861010638,-0.8745414749525157,-0.5083921304866594,-0.000202576053382498,0.08062359193264701,-0.7117723976711307,0.07631082273807317,1.321875665252435,-0.07638703134813185,-0.2049988074670486,-0.2244916121733854,-0.6947184095898054,0.008741538442545433,0.4010032164351505,-0.6696353443077332,0.02121545908642978,0.2191319417301897,0.2014834138441221,-0.4083404628163413,1.630128411728875,-1.043817245953196,-0.4011097667723566,-0.2300960444010652,-0.02806295809666794,-0.1560197710527357,0.1098608847019288,-0.500165191041395,0.05176454004532009,-0.3379492585183164,-0.1245466919869712,0.3106798116159699,1.958890817235552,-0.3259715667686267,-0.6944843974631322,-0.3451320897081282,0.7016749201675033,0.3534164931908642,-0.1548655688972249,0.3538937031660185,0.7843494226436937,-0.3024450896918316,-0.2310050970313184,-0.3271762986381309,-0.1005171884373814,0.1083768547579653,0.6571909442056018,0.5030637821861376,-0.2702260253497009,-0.4466505456277609,-0.1141835682473332,-0.8770998764543555,-0.5568071299604455,-0.6827256582183411,-0.403951069397267,0.2473904821266098,0.01680434734472503,0.9900040711307115,-0.9787113511969515,-0.1073180424694534,-0.5730666232171997,-0.4401507823572323,-0.2191564443508512,0.03383451925471689,0.8555404868067105,-0.1397300949968,-0.3915093790013758,0.4347475191975663,-0.1647360385637553,-0.6440003334146692,-0.2021109948361935,-0.4806073902856735,-0.3560252264810461,0.5625130718785101,-0.05224541142425514,0.546114123659155,0.1739566209031579,0.1994521509134123,0.4629818122573819,-0.530038798522068,-0.3257032471825729,0.04484278286340596,-0.04417631236858667,-0.3008733096143588,-1.091267006132964,-0.6036876494105652,0.1211440465091627,-0.08451091511332698,0.07069690531276672,-0.178944590227522,0.09643757725452497,-0.3694895841286866,0.1315937265799661,0.3091900176784976,0.184773964712025,0.2922618066297177,-0.1840562832930175,-0.2624920848970824,0.06113718058502569,-0.9915842458510218,0.6055690341169998,0.1876174874963256,-0.7095239522649082,-0.6067908395741816,-0.7535273566156365,-1.032016405655319,-0.924245327613885,-0.07775913903756078,-0.2786641794509997,0.06469598762328599,-0.1906175014215793,-0.2704430548965918,-0.3514429240469694,0.5917405783624148,0.4819626717241272,-0.2713562105072567,-0.7279880986865654,-0.4093579071559832,-0.5374240811525375,0.599175881520135,0.1705389474348019,-0.8116138279103121,-0.05323952812057843,-0.1700543560257043,0.1985796855721726,-0.4865721569646951,1.11974288441537,0.4845527261310928,-0.3134924941128975,-0.09894814275553365,0.1545315482914757,0.06792531543519362,-0.06796506756302147,0.2463607231796227,-0.3228339046281637,-0.2416612562351728,-0.07383302495567259,0.3732078795341028,0.1970646438963076,-0.4550379441587449,-0.3987286425152903,-0.362086051350607,0.2662336455466546,-0.2602104634548346,-0.4661635772418702,-0.5936258140802348,1.179090420646966,0.1827513263345918,0.1961539055656366,0.4153062436591075,0.4563521136332429,0.06349616106853112,-0.2190827071353355,-0.2526420230189776,-0.3332238430873359,-0.1810673328670776,-0.6704105851774178,0.0650601204807873,-0.2775723761505845,-0.2714832623708401,-0.2229621274895468,0.238808323900136,-0.148049741171912,0.0769666356562278,-0.4421207170797599,1.122813337073097,-0.007779557349865217,-0.4899832392278507,0.03425495459189689,-0.3699819151857637,0.02854602411481183,-0.1807289261021923,0.9943321589110089,-0.1953351580162906,0.3966371404560827,0.058931844155416,0.5396445102430987,0.1268964973666166,-0.01513536166003886,-2.314156681203851e-05,-0.4052640817577791,0.2635438335401278,0.1710064930785386,-0.6292819778048182,-0.006400410802420031,-1.192785325637277,-0.7617773938228757,-0.7899131444612995,1.083849884805607,1.635787870525765,-0.03772394292533916,0.8415132812060484,-0.07720239452510712,0.008494990791425568,-0.6726165206798573,-0.08402238099539765,0.1388051875623705,0.04371715033179811,-0.4336232345094629,-0.1081312689168372,-0.2687011322932217,-0.8298302729255537,-0.5462937160726478,-0.21047402756791,-0.4621178314285971,-0.402436394031815,-0.1993549438478699,0.2953885004765001,0.2288079163755655,-0.3648164226716135,-0.3510964769273247,-0.5367476513888769,0.04117916296732995,0.4096343050431387,0.1515561031374978,0.2773333894471871,1.340500306070343,0.6594495467494949,-0.2418116416132676,-0.4371390347872552,0.01176836327072798,-0.348050219807667,0.4686340647834464,0.09177234378712347,-0.2498134182067037,1.098193946921465,0.04215239126399668,-0.6128029380253661,0.01016176660273268,0.1458402343706405,-0.3572470235957396,-1.133941955199707,-0.4153805593856925,-0.1027765547699342,0.03227399012092436,-0.315937406423194,-0.1289527771742493,-0.3315688386766463,-0.3412258842523657,-0.1484572199449356,-0.277015979917097,0.04643232427860788,0.169144917560116,0.03017993212673522,-0.2583103133378296,-0.3041761507744474,0.7089381337597891,0.5817513667823001,-0.9696902430722171,1.80592809488897,0.05304778380820675,-0.1166689957041792,-0.433197148044971,0.2437925269484896,0.3076032566542238,0.4355353328241716,-0.06500266470674808,0.6096064009874712,0.2918109754509142,-0.04834337150751456,0.3314417545571858,-0.1186276736485416,0.09638024796575291,-0.8273784454049874,0.1513104539603309,0.09848940838337115,-0.2511228339870868,0.1008075706943991,0.09989849597769325,0.6739908567033145,-0.5843669161780073,0.5618325919935503,-0.3218310904407068,-0.4015834220569959,-0.04321097800638842,-0.2884368816032222,0.02202830137883631,0.5479992139720432,0.06351978664731757,-0.4082622609647337,-0.1556312306000063,-0.6454084003175089,-0.1884488580447556,-0.5760792354852471,-0.3010017955099774,-0.1004271542336962,0.5612615925000951,0.8491955789279076,0.04901273669242009,0.3704799268108842,-0.5270231806359617,-1.499040778412573,0.162215303055068,-0.478692193163707,-0.6502039191262358,-0.7109111803023468,-0.9252958925288648,0.2757893400862201,-0.06134485676898445,-0.3265706928006369,0.2352636755773265,-0.1500244025841706,0.5207645714516096,-0.4456718458295165,-0.2527909507287112,-0.7096123401876884,-0.6967504972573328,0.650429570834447,-0.6052203275602214,-0.799458085781371,-0.9228114172813774,-0.2730379161353239,-0.360872409526048,1.154381449838711,-0.2406153281577702,-0.05257952104880968,0.09893375838282374,-0.1328110580665369,0.08871869991714328,-0.9000773171601155,0.6080067353315471,0.06550442870051539,0.03054526944807638,0.1394609732804499,-0.1841989482545487,0.3118841844071849,0.281427644726602,-0.5231341504031428,-0.2105967136430664,-0.03085057056748165,-0.1961542258452287,0.07938282739344081,-0.1225174632314199,-0.7285927721923481,0.7067186450505216,-0.854281024524572,-0.4112788948544118,-0.08491159094748914,0.1768667652370537,-0.4258482446057204,-0.1852986792887557,-0.6179095263848647,0.0483058573396402,-0.6411358909628737,0.4063213126729022,0.002884251379871726,-0.5532453957159397,0.6848100838221394,-0.2403281471497454,0.009957248695616523,0.1676609116885608,-0.1446664686854315,0.365828282123854,0.7091627435831445,-0.427460165705057,0.1782451904362631,-0.2450741441969589,0.2562961684653278,-0.182466696039374,0.1143753695379847,0.9252710725072547,-1.429589540022057,-0.4536541370904122,-0.1813350312496727,-1.231873199244349,0.08750989745164783,0.1377892372114998,0.5098035761615516,-0.007808342338778914,-0.09698888039034277,-0.3699620372313464,0.6117245993897116,-0.1270104199642579,-0.1145999034720449,0.161716060366419,-0.1571183185817251,0.425632687618492,-0.4053617398200707,-2.121409293745874,-0.1025054727949655,0.1000219350058584,0.2232552887113557,0.02423138411466883,0.0966361038955952,0.9058173512904804,-0.8473894443213444,-0.3748113670996489,-0.1168117536047077,0.0730088437823297,0.3190613091479056,-0.8383972196282867,-0.1217859713289272,-0.4965492153452979,0.2225328293977525,0.265610403711498,-0.4282852887205755,0.2099644065411889,0.5839641477350979,0.03388010696564354,-0.8912551520747114,0.09251629197865155,-0.3810320121817246,0.1865970930227447,-0.181415537762395,0.09078812891516672,-1.060741264785033,-0.3104240123840908,1.166572274985927,0.05453761880977261,0.1994215133691485,-0.1622360383319738,0.5401239892310534,-0.07926204242098438,0.3336380727136004,-0.4921666157131565,-0.1794011339190216,0.1268416866283643,0.1488568305858073,0.02722329034296938,-0.1346644589987547,-0.5395325171606015,0.1842802304640057,0.2313847909937199,-0.323388878512323,-0.3039917101701741,-0.4488660072993945,0.4547826965822248,-0.4007066671498996,-0.09461674820072445,-1.071647084984972,0.5697348347070932,-0.3472734438220057,-0.2870417232399108,-0.5388973396435012,-0.3286668761818702,-0.3432897934171664,-0.2586939857415991,0.8286069369502701,0.1409264989181212,-0.2912367028395803,-0.2079394606148371,-0.4463691144099733,-0.3596797178651827,0.1113834599427496,-0.5524918650181729,-0.1086463988764854,0.1336352259655826,2.025080053042377,-0.04217258452879152,0.2427374475602195,-0.03068490500856037,-0.06013855160801383,0.06452409871177603,-0.2412191797886071,-0.4039698985998713,-0.4989071715810288,0.9930561514899582,-0.1161866318000581,-0.190305333691308,-0.2219856201618161,-0.3317711387353866,-0.2672156606568162,-0.009286846402520246,-0.05218760811437009,-0.3819242069343089,-0.617089091972313,-1.000237888377712,-0.1636040448754103,0.2333037914609511,0.1476749479015686,1.030611958541652,-0.4411973712462849,-0.5656720295639068,0.6226264130792781,-0.05999505143594198,0.3901237211455931,2.100188791716835,-0.1118539799697309,0.3491375760069653,0.1194846185540987,0.5774729650233409,1.224958015581836,0.1295488639882159,-0.3525696983296077,-0.05994437420880041,-0.4527398361293677,0.451280601532425,-0.02276545900979815,-0.5003937412878404,0.1596910934636305,-0.6026052148613656,0.3845199379900482,0.05640216181622056,0.2292447792925043,-0.717157180610847,0.07139494476645125,-0.5273475639597498,-0.5106622194175773,-0.1970518799179017,-0.2678993862760173,0.1919892975498513,-0.3460323055171388,0.02053244646416551,-0.1618575227165264,0.7316761090734134,0.4883418117524641,0.2810140671337247,-0.6459508191951906,0.0004953585445137253,-0.1428213432125865,0.5958616519801965,0.7750978037813246,-0.02938595411790616,-0.2207825097208888,0.1465732799350256,0.1673397802956102,-0.3044837055178089,0.3580289832216795,0.03630688662015823,0.1357909865502955,-0.1004851177295752,0.3751439090751275,-0.9282582153884511,-0.1399330156554493,-0.5505589938962283,-0.3113598379217598,0.6865625062786389,-0.5527521262598201,0.4243147126135887,0.3941255121354502,0.2701995835142841,-0.1900404539761667,0.03334313278129458,-0.1762162958807261,-1.024999898202397,-0.1481605127497999,0.07512244248994335,-0.176151253217042,-0.9695794676578963,-0.07112880352700109,0.3535916276970048,-0.4072110466665518,-0.2345915309955315,-0.3585688855047826,0.3674565575997717,-0.1127761651300696,0.1970786483276487,0.3707537951997389,0.0560349490982958,-0.6782367479796666,-0.3475706751929315,-0.8905608079800822,0.2555356733833632,0.01403949961311065,0.4496406271852927,0.508416348974457,-0.01417024595188821,0.04041851421121176,-0.5557939738796944,-0.2297123183358743,-0.1568441063140199,-0.08150122543014106,0.7716350669157066,0.01797639624684863,-0.5534548728555347,-0.7409544833025538,-0.7712236357339319,0.4739989492423001,0.1902741942493898,1.294713482066213,0.1517020769867137,0.1338323232697245,-1.143371695959931,-0.2961645836310954,-0.3802819636447849,0.7773851884641116,0.0711426895315629,-0.1995856095499713,0.01577036819282353,-0.438043311709137,-0.3970187599983718,0.3314215134829798,0.3428399794391444,0.07326687794630872,0.08440479921800244,-0.6402967553292531,0.05365598317537471,-0.2517648903135325,-0.4796695137351417,-0.2885951169201634,-0.26558720737435,-0.1536288138259343,1.184877669978852,-0.7661660137095863,-0.5815157048756997,0.2355438180257756,0.2181876365192374,-0.5441913118165083,1.912271972452333,3.309122891784689,-0.6794828429628856,0.3219644997744628,0.0663146778640928,-0.05759344458674171,-0.2565090436253333,-0.3521676453492418,-0.5976154769014345,0.1878843834865251,-0.4117208330401899,0.1538257874601631,-0.3187399588127521,0.393520233393807,1.058869654905434,-0.2543519794817823,0.05353462995627792,0.582993909794593,0.3460709671322788,0.1006595752644474,-0.382302501764875,-0.5137656027135518,0.08678531288393386,-0.1709660528161839,0.1145841927123039,-0.02170947291581953,-0.07970071528914005,0.1113192500709486,0.2142265653527274,-0.3304845337674296,-0.7916238525737429,0.2442737129160952,-0.3075789796056208,-0.5105131105737613,-1.377503815275785,0.05185743474078527,-0.02634864737521823,-0.1513167352243447,-0.7991721924455639,1.447735030267534,-0.09232121470083321,-0.3878213366631044,1.867598800766518,0.2976171508630525,-0.2977318775321332,0.2374449136377425,0.1448551907629779,-0.8716598669951076,0.1584014301393447,1.323600874610444,-0.18159699041173,0.02807451695569507,-0.3312636412592169,-0.3339408359031182,0.1720179569564771,-0.4308132975443427,0.01312592837605392,-0.1057949929994438,-0.4725312381886477,-0.2488656237688553,0.1923750487691898,-0.1793214111623221,-1.257289013151045,0.08441683622079853,-0.9339080252431657,-0.0833652671022839,0.7566065153808393,0.2254253212005013,0.04133368032362944,-0.009201713916119904,-0.4207391000150354,-0.8288616347099341,0.07190125198747542,0.7878025165493726,-0.1270395728136896,-0.1617892857786689,-0.879750692450249,-0.5165720061294865,-1.20756341044553,0.008513518320446589,-0.03615327846925848,-0.3079733393437799,0.831361435443957,0.04550640332710913,0.4016583434926498,-0.1625339784616177,0.5159814173297609,-0.6490827689935479,-0.05339359186582376,0.1984206902257348,0.04144611586383995,0.6005858832983283,-0.09716945758033498,-0.3446770678325722,0.3003238875928188,-0.3896079526644289,0.8376868644851234,-0.7345968652425795,-0.05502047410970743,0.3120142567147083,-0.4387412022593977,-0.3678847664509129,-0.230183771696339,-0.4752828899607615,-0.6953741165367503,0.1712620473404365,0.6563088282853374,-0.8738440186721247,-0.5991669705095428,0.4629308602319796,-1.162618806144521,-0.4589865113370229,-0.376564280002953,-0.2610897121417415,-0.4347509089063363,-0.5757295386009916,-0.2464530626669798,-0.3176004425651883,-0.9099529122993848,-0.7224681254518959,-0.8729592146401387,-0.1842028470017078,-0.03784800232992545,0.8334846192392474,0.05765614642065399,-0.3551285113685741,0.9990334123228433,-0.1334574845750246,0.1389165747771605,0.1178985300455374,-0.1367344394557886,0.3397520034358977,-0.3648482619002806,-0.03834676917617562,0.04436629936334976,0.1068080769482972,1.102932871715837,-0.5223572698211875,-0.6914382602464059,-0.670156949327436,-1.177417136908882,0.3328305593782064,0.08896385864382257,-0.3956670617819592,-0.3953885728975534,0.368578450628702,0.2621331629341893,-0.623087807311925,-0.3355118135154861,0.1618631020583036,0.3623306562182788,-0.3221776675579439,-0.2651333448513816,-0.1141940769115361,-0.6396787687667468,-0.4942407227565074,0.09048025624407412,0.2595936376547813,-0.5715732748468783,-0.8682341822508149,-0.04862708413866242,0.1299118818098584,1.916217154568233,0.6385791484436337,-0.1840061798908686,-0.6166473056218277,-0.4494290462395401,-0.2427683807222037,-0.153447324363067,-0.278153674403893,-0.6394669137329584,0.4339525908000723,-0.3377926206734811,-0.5556444207631667,-0.1464524277228068,-0.2023004952529668,0.006553363673605529,-0.5873263736044984,-0.5283611778727372,-0.5451789165698251,0.07784999851813032,-0.06295311260782714,0.1605006156448147,0.4347066591962135,-0.1817797584120158,-0.2579602227822828,-0.288732115637062,-0.2264606041168391,-0.3160159521048889,-0.2907728939454904,0.3732458638816615,-0.839276512701558,-0.2700570813742633,-0.3577193780366735,-0.4867467571184301,-0.2036039064314386,-0.2449978790579604,-0.5691074314771515,0.2292273449092433,-0.5909376211292269,0.1366049252603997,-0.7294792024992752,0.44159609116909,-0.5200075589332437,0.2394061030645051,0.00678346341491113,-0.06085260876538053,-0.8465785941729652,-0.4917044412232816,-0.6425105821246643,0.3500845144399304,-0.007919201107851215,0.03002253514033824,-0.05107962582087259,0.7291511061011871,0.006697762528826293,-0.4778097503935979,-0.4102094999049147,0.1764670017950602,-0.1043708600389141,-0.2160412648784252,-0.5413704672877703,0.4595272708537236,-1.317963578677743,-0.1801046108827386,-0.6837782072917566,0.7499819761388592,-0.1503958168062513,0.6139455536668089,0.03778403263540953,0.08412780499180562,-0.4872031419463242,0.1115570071183228,-0.2572744788232426,-0.48898431814153,-0.4423326522845419,-0.5026762050584566,0.1173957066541603,0.3576434730110755,-0.163988970907521,0.1835722583664921,0.1306242531994888,-0.1692157820763467,-0.5792101301096672,-0.152847395231381,-0.5116998578292378,0.6161622646504726,0.2937862310631061,0.2810593643079426,-0.03773849269261094,-0.2805639685202762,-0.6097965673477973,-0.2883938572698594,-0.6106954290299617,-0.4626041492142029,-0.8953398419520063,1.547252611093336,-0.2545236509046802,0.08523446489467949,0.3455852754033515,0.2368459022169049,-1.271691341095016,0.2001043648060661,-0.4448975721871851,0.2756693351422896,-0.2253805284765813,-0.2086551064757083,-0.4210855454415246,0.6057377127666005,0.05854611585324572,-0.0972702564915266,0.2937533075996487,-0.2393904787676723,-0.2285930916251356,-0.5596789966154324,1.064060423014938,-0.08962758841592508,1.041485664297947,0.7122358662680625,0.02168862000560662,0.2015427424055699,0.00651939103632941,-0.006018346150973577,1.586682036607026,0.3390568054008775,0.4023764822867753,0.3883968731293704,0.03428804963390116,-0.14143844302182,1.35262394831483,-2.410723917872873,1.42472341264887,-0.3767462553874001,0.2688010356000088,0.9319101422647613,-0.104604908512548,0.01635267372695815,-0.3547153530461808,0.5267557038862064,0.03159254453514909,0.1912794855513507,-0.1505262466350278,-0.1485246966737958,-0.7362040529959833,-0.2063332726539917,-0.1282383420261993,-0.2838212816063826,-0.1417522352770833,0.04471988687439953,0.2095415884903234,-0.1510829794269363,-0.6122844336327276,-0.1607522638642908,-0.5837474435046214,-0.0942054067877357,-0.4230689307038522,-0.02608524906851071,-0.07674092236470208,0.2672809948188304,-0.3218579558397234,-0.05563013738065235,-0.2013888587424712,-0.3147476814333486,-0.2264657000414938,0.5390851152205026,-0.7242056319427969,-0.9096267929992744,1.084001743153839,-0.2298697936582243,-0.8090244861440401,-0.005600481122711353,-0.3711451257926952,-0.06430330552142668,-0.4211559830347365,-0.266167881038906,-0.1758581964938115,-0.2359080274642027,0.3775582176069195,-0.928003803962912,-0.01098128938761432,-0.6898963247369684,-0.5143325982407537,-0.7061261584564927,-0.1048114951318688,3.182710707317471,-0.06696828894518381,-0.09667048631314404,-0.06022201123218953,-0.5736550207992649,-0.2408537109085236,0.1209921650925762,-0.5359764091722792,-0.3490181181264935,-0.6352202771522057,-0.1052704873818926,-0.6444379478953751,-0.8402982953369083,0.2828141475658867,-0.1503545806571577,-0.4708717630279319,2.130162903647494,-0.1299776988537973,2.065381072701504,-0.2879756932636198,-0.4470308053094254,-0.5758021323699651,-0.4456873265977022,0.2494441676774267,-0.2772073848724296,-0.5026121246049134,-0.1988901309923059,-0.8950083300735597,-0.4560749118516695,2.149121923148563,-0.231639212026218,-0.5475395765000141,0.03062223674634634,-0.06565402576059622,-0.3386677016367123,-0.1722368233477094,-0.05700159937264748,-0.6163846834121716,-0.1771305928467735,-0.04093848908861521,0.2398684838139259,0.3369376218155701,-0.2377924681273581,0.5331487452512444,0.6378741555544002,1.066083563646193,-0.5337237994412926,0.1444974700887364,0.5568395808635942,0.6577944495651861,-0.3165408215285447,0.4810020110884414,-0.4164308311123681,0.7359329833512153,2.936619680325415,-0.1353014055664862,-0.0001380873914475512,-0.1582875801430524,-0.1405375142224841,0.452049050778171,0.4651520253243031,-0.5260264987469918,-0.4757267018845522,-0.3633075292908777,0.3788740353366397,-0.318531905655373,0.07573035890811422,0.3570094137718369,-0.1168240066229482,-0.2047946359645543,0.1697147760545812,-0.0996678915762049,1.568642430044792,0.1145073874099843,-0.4314505166441021,-0.1898268698216675,-0.5609808934540256,-0.1951351070432666,1.672365597802996,0.1071606485820373,0.691034054610961,0.7341223521454493,-0.1818474624453667,0.251228871071548,-0.3561739572995825,0.006968456555555645,-0.1970536328032787,0.3343918782590804,-0.1380944326683238,0.176457737890022,-0.7089890550002842,0.005865070306790302,-0.03141849652006884,-0.04579839615004971,-0.3446110317572113,0.2283286549507723,-0.6051994030566936,0.3460673932854312,-0.1721856177176099,-0.3189564047380944,0.1148249929157193,-0.1263127254305792,-0.1436131750713432,-0.005349076718788438,-0.4074382982600044,1.223937820328592,-0.8879395034954659,0.6001948371918246,-0.1301200932979856,-0.1859468118038201,0.1287956374893632,-0.1502810590956484,0.5542011347813584,-0.291188003886209,-1.153429017500634,-0.7076962124129023,0.3427257778801689,-0.3679267104931546,-0.0425175100972845,-0.1549215094989758,-0.9779972298056242,-0.9772122393902288,0.07215142809164168,-0.2488260951760881,-0.2195305057790224,0.2784961149966689,-0.1205073602466191,0.01569674082374796,0.08683623838689562,0.4229735805397609,-0.1990224227269737,-0.5823389248343228,0.258200835035037,-0.5919346787891129,0.01241587387203803,0.4450076059222636,-0.4808434131429091,0.1547876458402567,-0.2463747166765082,-0.8882379652961224,-0.05075970885791218,-0.02617488300453464,-0.2021247687462517,-0.06138342631357309,-0.5408817148700825,0.5085470925554612,-0.09379360689343548,0.3927333689816149,-0.09491513639337118,0.04744303087316415,-1.101152150891322,0.5499557615771401,-0.4126336401388369,0.1652070881391461,-0.4160148201022582,0.4423902937402192,1.139557279770327,-0.1831827234921466,0.3025294103908523,-0.1377686544132903,0.8303750392490263,0.3192402972610795,-0.1153948371709707,0.09347307804134915,-0.1619876185811873,0.3600266913855019,0.03318845770905161,0.1409754328419215,-1.145182752514454,0.4309701393121903,-0.6360041970563174,0.01956072690013948,-0.2652248690717141,-0.6576171991585508,0.6707061617805792,0.2603993372075667,0.4928181157266333,-0.1217047211286429,0.09649135744695689,-0.5382809720678752,-0.6027911793821737,-0.3793508011781916,-0.5507924390851809,0.7771984956884592,0.3201044260725349,-0.05692906373158598,-0.01016854154951852,-0.05567723340561895,0.1319009439628974,-0.1394105033008037,0.8608033102753171,0.04756990094620751,0.5060742508432676,-0.4340948893960525,-0.4773389511514135,-0.1441587599716621,-0.6264159839281532,-0.5193087794153974,0.0001515290252310127,-0.4273078476092972,0.3325619821423773,-0.01684312216193399,-0.2743638072933414,-0.1555170798141157,-0.8810529151574692,0.04451541890548044,-0.1949114992555075,0.02330719325700265,0.05627117750107816,0.1692715281501564,0.9706514536624417,-0.06867060258290912,0.749824820808969,0.02074777101836639,0.1680401944018658,-0.1765228092233218,-0.6065564495197688,-0.2252030280826433,-0.182195330046125,-0.1476654357083632,-0.3429475639849496,-0.3207578710643008,-1.102024355134258,-0.002029131459426906,-0.4360943978318013,0.3265554301760444,0.5545580802271295,0.03022132551434734,0.4865353960846783,0.2780707149129332,-0.4498146000050028,0.6023966912422452,0.1938184760153294,-0.1753434522261734,0.03447109616933625,-1.08970719326101,-0.446081917384454,-0.1326580213607113,-0.08182520156993263,0.08142506303860088,-0.2749323259831148,-0.4475116575465615,0.2019257647696114,0.006943068841962514,-0.6746528423770867,0.7310621833405757,0.7943700020487902,-0.07205999728478867,0.4300987466075015,0.3018584131769778,0.7060955438886094,-0.7399319518948325,-0.3304126956158184,-0.1436182001583411,0.1563755151342005,-0.3609986355083949,-0.230647005271312,-0.1511081162998415,0.2691910099978496,-1.002822294817073,-0.1917594866482909,-0.6745938400398268,0.4289180718179134,0.1491144079132246,-0.3419611330344817,-0.2928053311938532,0.4046817510894384,0.1433191780458255,0.132135701736952,0.001804372887048328,-0.04273042372948983,0.8919418620302804,0.7478830353632054,-0.1291020789700642,-0.5026188851975718,0.8562491411487561,-0.4508146666407423,-0.1254644427413047,0.4801297423041939,0.4262265655383513,-0.4221339586697092,-0.3176290313603467,-0.1400885149113618,0.06872118987341802,-0.5336450484579912,-0.2596721151431419,-0.3023552307866728,-0.1401619805652463,-0.22345367508611,0.1097351443328123,-0.4459583572841511,-0.02005118389013249,-0.1815760541977268,-0.253672573608657,-0.1233373298942639,-0.2729209130745714,1.115569835445935,-0.521239917193548,-1.4778409036876,0.1249153122715865,-0.616694275914081,0.02928643733076325,-0.05329626482822535,-0.1189027795844982,-0.08361918887377488,0.1221650770903893,-0.2169548143725006,-0.03875015015529229,0.2186560243915714,-0.02655637382482809,0.1084636742344723,-0.0531875430795859,-0.6014996395562128,-0.7390885696651639,-0.7791214272570641,0.1005223181883332,-0.1075653271132505,-0.2064886198406378,0.6081981769529596,-0.2461881126958465,-0.1272997477761433,-0.05350069546806196,-0.02138646252419755,0.4174102416379874,-0.2556392079337264,-0.8068551901904661,-1.993884360302385,-0.1730241129292997,-0.1532182466508092,0.4117740047768026,0.3992148195647363,-0.2560758211434279,-0.3996352767410994,0.3393506030000538,0.2796332908164558,-0.6207828410602892,0.1432607362571108,-0.1768832099418862,-0.1321916144702255,-0.2254197474376173,0.3190838017758145,-0.01820579412019953,0.7719071604661158,0.8616036263139664,0.0131962937005074,-0.4202207634617562,0.06793205562615995,0.6238888546587366,0.03504156093757445,0.1148579058184924,0.6565952796090361,1.122562194982038,-0.1122089958000847,0.6199042046905042,0.1541551946486301,0.04153859373152975,-0.1763278529327239,0.003290276323545857,-0.2707324185829794,0.8234511838985782,-0.583336772734389,-0.5120263544003227,-0.2710485638332097,0.2611234769592314,0.0224209704115885,0.02267053384671252,0.1781710477138647,0.09878184338479555,-0.3166863464660418,-0.2630399299202401,-0.2354018211825836,-0.3153840694376708,-0.6834039287174934,0.5301338251221904,-0.1615856735574868,-0.2935853932894315,-0.1522956633315183,-0.1788418124008486,-0.1606503410914011,-0.08496777526907676,-0.654239887145486,-0.2287976644862697,-1.022768946428343,0.2595011552527104,-0.0451248522967375,0.1458579098451855,0.03287346299124699,0.3704865506593741,-0.09070258175322071,-0.6178822879295708,-0.3771705704287427,-0.108202357775625,-0.007250339926441468,0.3998907923886832,-0.2275891103834049,-0.4688461672857017,0.368182101342252,0.07993987816559092,0.2776147534750663,0.06708404575118342,-0.7376218555383011,-0.003147664186707591,0.06921097738372302,0.2073458462370325,0.7096220619305202,0.6072884580940033,0.1136311987722988,-0.1575798646991868,1.475118360469026,-0.6920615064017509,0.1459034240769545,0.4675852289778476,0.617055421165951,0.1031993561222088,0.2272117784304067,-0.312495090619504,0.810063740354935,-0.6318945256305093,-0.156119066098912,-0.3185079456247536,-0.4470120751524163,-0.4005279092628289,0.08895793298755778,0.1093097680531433,0.1020048477133923,0.9376735773895616,-0.5749451623862795,1.532996971129939,-0.776014725215064,0.2175050578365586,-0.5466926631900754,-0.1030756509974662,0.2311747819475688,1.535766456377875,0.03449527827343459,-0.03996870226511951,-0.1526737435002453,-0.9698180870854098,0.07703567567679501,0.5298627235913219,1.321459334323353,-0.243329678355635,0.06400752034301287,0.4474392142937628,-0.4403406486833028,1.387845903708293,-0.2041506885538994,0.03458289451371733,-0.0489487873525432,-0.3855956509969077,0.6387220635901414,0.1070739644470779,-0.2534785808520334,-0.3457480910454711,-0.3695356445977291,-0.3950192484905711,-0.1583021668606182,0.3202445030482339,0.1573703551721498,-0.08548546290056813,-0.1855655243520943,0.7128582263821087,-0.01538689617960427,-0.351052891039713,-0.2902335226689125,0.1435886465734488,0.1184044766898915,-0.4451636182185152,2.598179778972811,3.786373240566428,-0.1422991284315004,-0.2951437804266624,0.008022661798667593,-0.02895291740806159,-0.1083273257570929,-0.004096691281108546,0.02489851878788625,-0.3608063135157736,-0.09345955076466374,-0.03589734147725979,-1.37356268041758,-0.05006668056496589,-0.2697507896430295,0.06267038999887156,-0.38127579186731,-0.397220999652789,-0.1256544607943963,-0.07179915367160089,0.2641446220396742,-0.1552150126986794,-0.02776258604376995,-0.3333140861850595,0.07263494338793035,-0.4558803277205451,-0.308112861074379,0.03913281345071799,-0.7521696136918041,-0.2718140793795785,-0.3204283298113163,-0.1110942993227966,0.6493603997806892,1.187943480028951,-0.03570329865638776,-0.0728855870981433,-0.1165126669024088,0.07496640522534791,-0.06414136664607545,-0.004250067110790632,-0.6583921487835642,-0.1916695385899848,0.2352897157499076,-0.5372132269659817,-0.363413878065654,0.8468374990965768,0.4695646052087553,-0.05444774125672461,0.4057367150810833,-0.09579540849796538,0.1988349276388653,-0.5527527103283383,-0.2576042594733256,0.06447161909162469,-0.4329130767878434,0.1810807492939535,0.4267220235564307,-0.02833906907213954,-0.2865620978073964,-0.3813943929758101,0.09309986933858881,0.2506912611467333,-0.5857648678925421,-0.025281145057841,-0.0342850212539266,-0.5142307606854758,-0.2172561124883551,-0.03306409984181174,-0.4180040948725716,-0.2958482072674137,-0.09451239406822592,-0.1795629568501917,-0.534240324182192,-0.4413660044515562,0.01687510638857341,-0.2346235243852886,0.4098864458579853,-0.01389373042721323,0.3360167008530059,-0.117870228725718,-0.2791858281760305,0.4593116275671678,1.306581874113923,0.3292463206906622,0.9429671557934293,-0.7617023312898846,0.6603875679441548,0.02522246107038306,-0.1193489003483516,0.1599878557313977,-0.01343698444022933,0.09158861528822956,-0.1560627199325682,-0.1955846171867402,0.02061982830493809,0.01236334276901739,0.07904207651233014,0.1340814369566133,-0.221359065693771,0.3153554424158913,0.09056769181771215,0.03359692858278119,-0.7053266132140809,0.5065758282566619,-0.06176895536622123,-0.3152559161708156,-0.9395201867616446,-0.2548853542501177,1.784092875467948,0.168576518023479,-0.5137539972384988,1.326966949874929,0.624538328914405,0.6179027954597469,-0.0002554989091575772,0.2881179684861273,-0.08816924772825756,-0.2103100183857587,-0.1408027189506481,-0.4280860448957367,-1.078985139169279,-0.2438617753786095,0.2856923803688226,0.1726746116391917,-0.08238868282395607,0.6389959008211381,-0.1572687157222939,-0.02162472811662786,-0.1827305363872822,0.4105056419340256,0.1234584453754832,0.5584875965274595,0.2315461085580165,-0.743705277948332,-0.3307011611938477,-0.3595257348375943,0.07666456234637298,-0.1940487232748809,-0.7736642895937821,0.2749101273520076,-0.04770552984180076,0.1650597116596219,-0.2405138449075414,-1.205517785396145,1.214804596676935,0.6822224634846381,0.07440823137679983,-0.4170008036280897,1.128877056595261,0.4197946652194535,-0.03476678929490852,0.3212720281751021,-0.1358406291130158,0.6354434041286733,0.6826402418084346,0.4524915744399609,0.5243523423404935,-0.09474226385156265,-0.7861508304151098,-0.9056294643774312,-0.1196616496921716,0.8390025186455592,0.3989853903328471,1.667050306057943,-0.181251120472593,-0.427060797384284,0.6219151818505817,-0.8842421761972118,-0.517683532166872,0.130090651829973,-0.5913150991029016,-0.3500619751560783,0.2191790058743098,-0.4226111821062944,-0.0780036009772174,-0.4694244699809123,-0.08617520420290428,0.9608952194463153,-0.3171054401445715,-0.5886027044084086,0.000742988723568772,0.2752331438913319,-0.1084135492799762,-0.3414650413954114,-0.2583159356875823,-0.1892925447670539,0.4394910341178377,0.04715356462729499,-1.092395380489591,-0.2574418482503509,-0.01098423402857785,-0.2037163781035607,-0.5220420557890795,-0.354886367507541,0.1701147520343654,1.270622305980202,-0.1783066948316172,0.7763679479507918,-0.5700102516994577,-0.05333104341586186,-0.3772453505239879,0.4856936116494598,0.4336006165472153,0.7363398837355046,0.2937975222257914,0.4742681516193321,-0.3939970696863952,-1.130999591968095,-0.1770820862965676,0.02983379329941114,0.5042324359830753,-0.2561784671216206,-0.0543339064452486,0.07950592199910052,0.1634194037426863,-0.3231968405426657,-0.4937220607355959,-0.3932714469922908,0.009338461108501565,-0.0334954514214877,0.4589517881463412,-0.124052112037074,-0.3729084108232404,-0.4979010067492924,0.4195126704404193,-0.1011593022387949,-0.3228160337683229,-1.039360654680316,-0.2875561382395772,-0.6302264038570768,-0.1028105818972919,-0.04551918948861725,-0.4768053700584738,0.06401889700927887,-0.5073189698566419,-0.1970826869367677,1.030462225269142,-0.3987762518491863,0.2346178525292738,-0.4207018312517335,0.01774350278069715,0.5196403580121175,-0.3845943088578346,0.1498850704351984,1.273091736377634,0.2455436669190256,-0.6190796970436578,0.3004345127851755,0.07041391878657172,-0.09175442777474196,-0.2095234595801622,-0.3855162273962051,0.9020982080285278,0.931144468461683,-0.8992675079032313,-0.1860862604710056,-0.4225904009365376,0.00662295244537175,-0.2453205607904439,0.5571595563143759,-0.6981804386665394,0.856226240260562,0.7585303568934734,0.0785448800672594,0.2126612657001823,0.6005568865929377,-1.21786115460838,-0.03393862532266719,-0.04397041346481429,0.2428414430866602,0.189335678257595,-0.3848506874206044,-0.8142094417793674,-0.05952259570570872,-0.1998378362670093,-0.3638599528305889,0.3959588105496494,-0.2612217422638345,0.01277118858296166,-0.3597074468128231,-1.226749675197386,-0.2766333479553634,0.3387225353860367,0.7989347219950602,1.023259288755316,0.07212825293021219,-0.588040601884559,-0.3679714694501563,-0.4295220059368764,-0.1506850558000654,-0.3873963232553412,-0.6467555683264118,-0.3097476407331085,-0.350647071052469,0.1312795525091175,-0.1806048508672975,-0.1989652008184281,0.1118049211342512,0.1507010493489139,0.6603690503011626,0.4080725337286916,-0.2428709202224079,-0.5156025316538581,-0.3188580094754766,-2.256069932222211,-0.5429412666232709,1.419182152497378,0.4328948330709136,0.3838056285667383,-0.1480074072576194,-0.4662934839789247,-0.01536322280709019,0.6360552846435488,-0.1098595243455558,-0.4772632151496281,-0.1408698154909437,-0.7046670142286318,0.3185459343701353,-0.1496295489911177,-0.740928013722145,0.6646054708614365,-0.5459535804917662,0.5743623328190643,-0.4203208096059497,-0.4137737642583238,-0.2757278848940979,-0.2394499452646254,0.3662272394914567,6.111116285493083,-0.02957277897097205,-0.4637163303180405,-0.3011817323731369,-0.1026405768336139,0.1216455863212192,-0.5085521703812897,0.1002370705357019,-0.6728821098252001,0.7171049654721382,-0.05837543405939759,0.3143342369535171,-0.1875414585090643,-0.1608434809872113,0.6545821029404397,-0.4138998361980059,-0.4691715361556194,-0.1250817451692054,-0.7153197739314195,-0.1548940830503143,-0.2218916804694879,0.9069460530783837,-0.5440014466417574,-0.163358069548146,-0.3090390755583508,-0.2909631935410165,-1.34339778949457,-0.01589577355786649,0.2519901371182828,-0.4585321382834895,-0.1604998060901064,-0.4011441071806005,-0.03627153814630913,0.4260438580251691,-0.03386583949978203,-1.000589658638823,-0.3093113039306772,-0.5816221752088991,-0.0805284812587917,-0.3051367520998032,0.1295721408561019,-0.04048631953644583,-0.4339645309387493,0.0737095256817098,-0.266280388972602,-0.2838442876460134,0.1576584784526906,-0.2625394881081862,0.2974522831958383,-0.07279455445340272,-0.7164129363183407,-0.1973275096565986,0.2011028618969343,0.06541911080421688,0.2032975773481663,0.2675009762468176,-0.1715189994157455,-0.4157499829895932,-0.3232160786127159,0.3225363425684072,-0.1176158552672996,-0.1110530230086045,0.04742723654061774,0.09599985005217303,-1.563858062192662,0.6210796852507072,-0.01299469229195506,0.3291495666739979,0.1782855183268824,-0.5553124648792243,-0.4164422345132494,-0.06518418102863055,-0.3109647943617663,-0.4341603115060242,-0.3466047755385901,-0.4210164891588039,0.8965306439615677,-0.6360166266414592,-0.5412841094288658,0.8337974584569994,-0.9500557973734355,-0.1620665432638867,-0.482219567523936,-0.3307294649954599,0.05524998432851889,0.2752321098319767,0.4467299746251885,0.8531108633205909,-0.6120978298594569,-0.7003020199465924,0.4103850398292074,0.1727121242913862,-0.1965495713485083,-0.2972101798074896,-0.6604055445890553,0.0001495460108939057,-0.3115157055397197,-0.4773097277998417,0.1978087971540543,0.007750584481061956,-0.3961231441360705,0.239896565829603,-0.3223596218833241,-0.09229637470473238,-0.1902959245003289,-0.7024988419702908,0.0125919767046969,-0.04844356870547748,0.3841210492758351,0.09854910959950605,-0.3447566443515537,-0.02106561661348897,0.3582046008822835,0.02295677810902703,-0.02021817626106362,-0.2090398997934231,-0.3073364828126972,1.200784886777096,-0.6665140061941542,-0.4346040113743387,-0.4721029716731248,0.07215424166881325,0.07229178265699548,0.1316676898146324,0.9374795678523336,-0.005041719701290748,-1.042196445460062,0.9639636588789985,-1.706891270797305,0.7548584936944622,0.05118353587296408,-0.14156309272783,-0.1957224282360913,0.3797849920493109,-0.5660385297071209,-0.1997289018339828,-0.2011619598667802,-0.4734655014010423,-0.09954564263771006,-0.1211584879122585,0.9609251559650497,-0.4159380026314238,-0.5703510744327408,0.3832080005028716,-0.1657958129482781,-0.3948730989039387,-0.2458646715956456,-0.05469269512888923,-0.4277750412486372,0.2092201643703166,-0.3235680706001755,-0.09906925738926727,0.7554898286336245,0.118389750865779,-0.1653882372869995,0.1858252035445527,0.06070104998549214,-0.1344417220921245,0.6017038174499415,-0.205431875561963,0.5222969707666444,0.1598152955551599,-0.2103729895332347,-0.0604060613971794,0.0001864250667443311,-0.7418552734296781,-0.3327093544808259,-0.523676599232726,-0.2294166710652891,-0.825247859153177,-0.8489839030367885,0.3052039893601988,-0.8974131601403278,-0.1380680821078508,1.180201598855703,-0.8178333311801276,1.716670723233302,1.008221224140672,0.1259048669616058,-0.1606934663532229,-0.0608570809851417,-0.2036777429646357,0.4594349741903744,-0.2270455661577336,0.306260511698789,0.01126315192210116,-0.6694577492723967,-0.09375437224130598,0.4499103767314061,-0.5185789318656256,0.2945014351305548,-0.2660223520196087,-0.2240424664650204,-0.53506062271008,0.08005230675332363,-0.6053058743130765,0.6968564704946527,0.3902467305709385,-1.051237345016353,0.1364348438459349,-0.1681154879910752,0.2500682331524556,-0.3849504077003741,-0.1791380768626941,-0.1249467350479901,0.08363918879947156,-0.1734752580098196,0.3061754641549064,0.1524415371283172,-0.3367532818198712,-0.02930868482697042,-0.003886454080669407,-0.9160390127215779,0.1210598207397438,-0.4561683608347913,0.0729217794920142,-0.2430837124330276,0.09078360881842562,0.05893588286928061,-0.1090835138683239,-0.8278238473762729,-0.2594277014402959,0.3355446780887497,-0.1439110655646052,-0.2567751093962707,0.1821522509104557,-0.8406992977893016,-0.1437393764643852,0.1624110646527124,-0.4071782847156667,-0.5438374636806276,0.1551884951659488,-0.1547878188319202,0.08672686569114346,0.8497933272426659,0.8514564180995092,0.9007993788347585,0.7860051517029445,0.641865339254817,-0.2129881758893271,0.01895518603546934,0.4080498269976067,-0.1842725565404507,-0.3724788360494072,0.4849101097900612,-0.0548048533335762,0.1399608860107342,0.9789647340073586,0.3014709803884036,-0.2662614222588918,-0.03178336030905576,0.2780971634034891,-0.4493693019313619,0.1809626626015211,-0.2618695492046215,-0.6672285874533479,-0.4036076433566612,0.3361428886883846,-0.09901261752254566,0.1340952300965661,0.136879399991255,0.3251664925234108,0.8836698282832066,0.1911721101183054,0.5827052817854785,0.3568865366990188,0.1329662064891813,-0.1568016324517081,-0.1792891718080341,0.6288576691489616,-0.2235134285050407,0.3967491729294986,2.977580377176026,-0.211966823983748,-0.2830284059814823,-0.2674723235141985,-0.09507583864145737,0.1688475830779988,2.252280557211119,0.005244952989261121,1.163556722842057,-0.4504051334151072,-0.3888704526160374,0.5253757369461834,-0.7087138047651632,0.5491594198256937,0.885793173976977,-0.570129817111736,-0.06614926785112636,0.2400459702102299,-0.04837710075261728,-0.2509725137228365,0.2191866515441404,0.158896046386153,1.258065768069672,-0.3024647866697455,0.3555007834995929,-0.4014902238690469,-0.5263739249389229,-0.1330940499247749,-0.06062212498676398,-0.3956281817074409,-0.03924890858094983,0.2893915177860688,-0.5186189991553943,-0.1303908496290798,-0.3887509667493476,-0.2280969096921515,0.6503452455594703,-0.1418471349529003,-0.2928095222507777,0.0009553763784954349,-0.1753116176398983,-0.1661067302528517,0.7432830641945736,0.1955656472977347,-0.1527710501659653,1.533991329225625,0.2427716362033782,0.03475059236824082,0.5511950261553356,-0.7917834013647542,-0.126781468767571,-0.9351354095870327,-0.4473314960580098,0.6714530440329888,-0.07612942250491127,0.4841266024019641,-0.2364701716671462,0.5237214504907345,-0.2538284152241799,0.1466744431970123,0.5845884719473824,1.052769253484223,0.25036087974605,0.1567314007295136,0.8950173398159577,0.3456326385973713,-0.3864924593664654,-0.3515008810038269,-0.2124308176328552,0.1317848938083663,1.441472133385173,-0.007719235790553496,-0.3543025554326268,2.363705737136467,1.61337410553089,-0.3086317358651038,0.7965831969050077,0.05049429315676492,0.1861798856915172,-0.025519185611784,-0.001970593643756101,-0.1673490692378457,0.2138691620511091,0.9833896169137606,0.3526399001378592,0.8808198911607127,-0.625438606087337,-0.3616288417136038,0.8918112292303795,-0.1568213611825449,-0.3578408610402802,-0.1644963766260911,0.07569216335147365,0.5271384750303847,-0.8699352739549809,0.7210310456150061,-0.2047901824833125,-0.2501952625150208,0.02937242172065336,0.08762307124179372,1.29920285993795,-0.2112492987592591,-0.1351191767428228,-0.2793443095547974,-0.7211400969409146,-1.487886044288806,-0.3795189722877639,-0.138533150494781,-0.03059820348326085,-0.4312465367577518,0.161875655537175,0.3661236726509478,-0.1053424000538775,-0.2563763450636447,-0.1358376278169645,-0.4362082498871472,0.8062265711642334,0.274251806506641,0.6379970509936925,0.07493616464875934,-0.1241320255769584,-0.2290071264330739,1.118184996795506,-0.2377065836687815,0.2391324717985663,0.5385491735081726,-0.3056972386806937,0.2714905888676711,0.3224564847765241,-0.005013184787321845,0.19275417162119,-0.194557060646206,0.2140902697534691,0.2112714722462369,-0.3658641762278446,-0.04141395461363506,-0.2218829374905049,-0.8105902919803872,2.469424947244213,0.1520180080804536,0.01757562147241368,-0.02399730007713943,-0.01082846283078126,0.08503861348512556,0.6958709974870515,-0.2284348532321856,0.8159275485170755,0.1190098959819913,-0.2632933225360026,0.1681896738916923,0.5603940488405347,-0.01008021175786481,-0.3526485561351649,-0.1577447075319988,0.2029873550450979,0.2480581575741657,0.07004405143355022,-0.02019401568308889,0.01162934285387346,-0.7635199375529749,-0.4955833055716742,0.01726000031683841,0.2440989097380421,-0.4665807111656583,-0.4073718020680295,0.1569917812511333,-0.3824799270303683,0.5711928455917464,-0.1124578071368366,0.4154247765786374,-0.3035233222709565,-0.6614922102610384,-0.3333591193376852,0.00570666694977944,-0.6341617105702521,-0.02401554656484825,0.506452151625241,-0.5972457526110582,0.01970162006575344,0.1282825427857653,-0.1346756583139529,-0.118366656600982,-0.6873655135761385,-0.7333098870765697,1.974634794861051,-0.7258494420530205,0.05289075200298909,0.2577259165354678,2.851780826413276,-0.4654712984292284,-0.1044225578648718,0.2054722736122335,-0.4159406211375076,-0.17551317157416,0.1018807710705684,-0.0955532302654605,-0.1302663217654683,-0.6358567105972683,-0.7807399484958044,-0.1654616322507334,-0.3789944008187149,0.1543966891255538,-1.160442120335551,-0.3692998309452832,-0.09550060026153355,-0.6144546265005711,-0.2907819942506583,-0.5630300595033058,-0.703476076462083,-0.1760439082865184,-0.3507642434077699,0.2682145817101053,0.6753420552249427,0.5138352297054889,-0.2023979925043732,-0.2052962293362944,-0.7425873038569436,0.07892297807358693,-0.1922743288842711,-0.118802998055203,0.02780706498727326,0.1440904107032286,-0.1915531944594369,-0.1846707970956116,-0.397193665633103,-0.03298741711520234,0.3045773644286043,-0.3332817277133114,-1.028551132975046,-0.2166795972842059,-0.2051977693616689,-0.2057576070721137,-0.1955771365594085,0.2433900152264123,-0.507817653297297,-0.3045804976683504,-0.121304779881314,-0.2364079036617073,0.02116086708701793,0.04113990672041658,0.1356117377596595,-0.2974319676560137,0.2892283355274506,-0.5283012047666562,0.06624287235409783,0.1752433650401994,0.2069110316217633,-0.4907716424032783,-0.1395660527595217,-0.2246066426440748,-0.3479264066800874,0.2975596794069035,-0.09124116894957029,-0.2203147276764204,1.174060187139523,-0.4465370929208385,-0.4184945590906616,-0.1534588774876564,-0.4023011008400723,-0.5367253952376556,-0.4514267140861509,-0.4501669903821611,-0.8657248517967253,-0.3032365939317914,0.07123361574596875,-0.229948470365568,1.151150953810881,1.51463797473542,-0.9971613044200318,0.2111075047915778,-0.3263704085423749,-0.1385747562807189,-0.06817984018279448,-0.6734107254157119,-0.08918477138903956,0.1130172362171873,0.1503313842014233,-0.305232534910554,-0.2639394916232057,-0.6301964616392409,0.1012397059274743,-0.008784462829868759,0.1806368567021343,0.09501388092106985,-0.08360907949348187,0.4498519142431619,-0.5604323387600881,-1.109218012398167,-0.2609432234414547,-0.7643108538544594,-0.3887206848035767,-0.3427411883296551,0.05509878461476171,-0.5260973904267124,-0.5240059160729575,0.1234152145176968,0.5828671987917451,-0.2276470838857226,0.1789242372591101,-0.07631061574784376,-0.6474311448830996,-0.648647529052489,0.03190239090581055,-0.6169268576531749,0.3002698527379549,-1.147109610463728,-0.493468335482471,0.2472380865456658,0.1676199656862742,0.3630549352421446,-0.7070076278643248,0.4970180119079004,-1.16763498902011,-0.3780931287415294,0.1224433110286196,0.03538777244525932,-0.03336855594674871,0.01357261984476498,0.6502520321764239,-0.5050381671492271,-0.1201887239820057,-0.7525960039965397,0.2115135333792326,0.3546374541676319,-0.3346768412953957,-0.2017254971616147,-0.1041485976731187,-0.6341383475362486,2.768571802955925,0.013936155400724,-0.6151383209583955,-1.053291541024944,0.0507688233910663,0.2878168199367173,0.04893064785298328,-1.048900001689587,0.5982627867724996,0.02556159447038268,0.4596144677300444,-0.162858648762834,0.1204028598662535,-0.3441312921980755,0.0007840711627723346,-0.03879039624146519,-0.2100893141603941,-0.5368168406060255,-0.1864346570174455,0.7901597440770963,0.1644732359801245,-0.6613213492871248,1.881417289249793,-0.3192573590418433,0.03825736507921431,-0.09717573187656249,0.138689313338729,-0.2536118507334333,-0.313846193193109,-0.163936553462503,-0.3123386095056843,-0.2157618290745705,-0.1311972343379209,-0.6130531616580065,-0.5206396118753305,-0.4667325540388492,0.3162087038308284,-0.8345135018435054,0.7048778054542425,0.3935868141559932,1.180488445625231,0.1910890787274695,-0.3282855635850295,0.1169705553699846,-0.4195966762682795,-0.0883536754306075,0.275458053096053,2.269433906635628,0.3367725951868468,0.1609879698170064,-0.6482944085977438,-0.4678430239402713,-0.3456223067374548,0.5780770300202484,-0.3803916892734974,0.1294230464871083,-0.08851099767496183,-0.3959994431983747,-0.1008604966324936,0.1691482180102415,0.07738189670107833,-0.1179806237885897,-0.7068583941945306,-0.2966552086810296,-0.5130265560153245,0.1209674450345711,-0.244464453062336,-0.2304077346712451,-0.24104411304181,0.1515946551137699,-0.4820710287689569,-0.3922718129828775,-0.09176857363397938,-0.5258237820941837,0.0647880515378403,0.004515964966620914,-1.423420598515241,-0.3075471122056396,-0.07898562242722321,-0.09083340756306961,0.1537083240879563,-0.3224374134993866,0.02044580832993315,-0.003045316377546541,-0.2752060876765282,-0.2558929779643304,-0.3128572199550232,0.4337734324054473,0.1309201678971033,0.6074361450777871,0.8089153603613947,-0.1774220032770783,0.6663314000757132,0.08490232297572621,-0.2343371209006018,-0.6162210558189805,0.4982145111802267,0.4723088679198272,0.5719161018332027,0.07888024774285472,-0.329171021167606,-0.1384747470228334,0.01013508486421872,0.08457053141421325,1.364948613178256,-0.02641643948620242,-0.1034558453509915,-0.3806201530407645,0.2769229738151152,0.01682366702183562,1.591758395618109,-0.06653193765018496,-0.1236080054475602,0.3696126514180665,0.1254905216753405,-0.5668020435571763,0.265487273410054,-0.6766357395992557,0.04420120801443808,0.6199788526621406,-0.02055069397339748,0.2804870353506203,-0.2485431960905775,-0.4910326798858244,-0.2505498561266744,-0.2468700684529497,0.108807297826031,-0.2274780611426061,0.8245551634177667,0.02950526789323624,0.1538479699971584,0.2372891339938285,0.2111883633886474,0.5633479374135254,0.06544495419365803,0.0009877126671859698,0.4820485779293829,0.02968401949892655,-0.2753382646298786,-0.2302019123230919,-0.6460915715965206,0.3795125410924743,0.01094019996675775,0.05475323340418502,-0.04107810311220957,-0.3259819293830752,-0.7011605733423311,-0.08162533872481646,-0.1141515765925907,0.06273973478821603,-1.263062555862151,0.0339687437357495,-0.1626969746179871,0.3835327632301836,-1.096047114424263,0.01911297620076111,-0.003593845184268646,-0.08367715559443249,0.3226340680044387,0.07265474997245686,0.5509411450454759,2.939583097312046,6.692636866732733,-0.4917066889917888,1.692016406388163,0.4767563751678212,0.2688128590718983,-0.003860694354503642,-0.1840338869259796,0.1332978736746766,-0.0238380606118123,1.184502932815782,0.1776871484971949,-0.8241297100583184,0.2626068817480673,0.1096826231137232,-0.309821836976559,0.250021475831555,-0.1643214337305786,-0.3988487640960248,0.6015792133792515,-0.1040694562620356,-0.01310278392902732,-0.5396323861021988,-0.2541334848629893,0.4376135867868678,-0.2379747158666792,-0.8409634023392365,0.3139264537377094,-0.4118929992283803,-1.158649913977067,0.5844065766574043,-0.4812051595602495,0.2366148311954283,0.03926055249789555,0.02777815331064847,0.9271845066228088,-0.3121580552960143,1.240141374416135,-0.08145019528028283,-0.2058845692597452,0.2857351029157464,-0.1867598835679512,0.1899240493343671,-0.08778148909737339,-0.5380249400400743,-0.4876061365186929,0.08104982713333034,1.63542244550725,0.08146370960949584,0.02488176331238892,0.07760178239556664,-0.4179462480286394,0.10710719316627,-0.0004143663931247243,-0.299112769447093,0.02442675285324554,-0.3502725319158536,-1.188159215126105,-0.7265921788632521,-0.180108301033958,-1.293814531494142,0.03610177312622873,-0.4859371306330874,-1.181860977072036,0.4235569415513979,0.9502807595476217,0.3430266185666442,0.2446762217707695,0.3925153063034771,0.1566443365665928,-0.4894908195741184,-0.265832319564828,0.1471326663609352,0.01693583444358285,-0.2564281573961955,0.1957018542171791,-1.180962895009605,-0.5900614610891949,-0.5844763911338122,2.034448081404477,-0.1719799116398171,-0.4599013960940187,-0.6210880431860976,-0.4078347341403087,-1.012539802165403,-0.04002791494070739,0.3406893118485403,-0.4533561800554905,-0.8889152765801039,0.05180637728350318,0.2096803481394719,0.875967415408897,-0.6882066543128104,0.2623288099378895,0.03932584986355124,-0.5934523952736972,-0.4876575185553022,-0.1460732797744602,-1.79563728670168,0.4620618523511259,0.1240179022372329,-0.1828857949239167,-0.5259567817761964,-0.9669618223372686,-0.5354478769945917,-0.07893513801400963,-0.2007409821723795,-0.6290642416749812,0.2708665055414536,-0.4811882937268838,-0.888952314941698,-0.08974877447763924,-0.1028712096448832,0.7212509956351465,-0.7376081483320373,0.2079643266967335,-0.4854905400813195,-0.03973087629164748,-0.3504243245329762,0.1324448935834058,0.3072878245694826,-0.2956308409396238,1.161884216795663,-0.7829207748854755,-0.3046135496854764,0.2204766313354521,-0.3749214127066445,-0.395050134496257,-0.4718668165447713,0.1747472281423725,0.7369358904219706,-0.356312330335646,0.219177316589594,0.5865380549008067,-0.3004142392815158,0.1547307973285469,-0.311043919500637,0.3027477259955368,-0.2901885956653922,-0.4949540343569253,0.09682310244718113,1.342051245130236,1.528215903853318,0.07683776947600772,-0.09625218128531826,0.08604645278091606,-0.1643190554344578,-0.3529292573549198,-0.06554060865235939,-0.1490274990245918,-0.6609500092833028,0.2590793583518256,-0.4204809612401912,-0.4629686028597743,0.4771547985699386,-0.3407381306863669,-0.5700037822232427,-0.07865750872465621,-1.085496629298073,-0.2542170045127381,1.353546041421668,-0.4041936771759505,0.2668788260726671,-0.1528863603318062,-0.09573689146686215,0.3477412181322433,-0.3950081283874372,-0.1842558774200253,-0.06602607532533908,-0.4830934085498873,-0.3324986485214818,-0.1784797711037766,-0.0496125075172609,1.260748769448896,1.026440239376231,-0.7229837092268948,0.3127622804953125,0.4364724871575279,0.08230669064384283,-0.3171348157970724,-0.6429997412140369,0.08218547298628689,0.002268460183535523,0.1590888696712212,0.1796315988762766,0.6112804105037741,-1.014497258640306,-1.01045365050332,-0.09459247707432332,-0.0989536046249955,-0.6363253038955878,0.6952943331077134,0.1841703532194511,-0.1134961559652553,0.6360500439855283,-0.577340764154157,-0.4881134907393065,0.2078676376208977,0.02306627908404198,-1.249266808295608,-0.1682222659000498,-0.2250262619428974,-0.049409986767173,-0.3807308378825073,-0.5473640736224461,-0.03526122327176089,3.179127772588719,-0.06957918063046979,0.8384993343683337,-0.05056321134038681,0.534654117509903,0.3511194930176076,-1.269294213295683,-0.2381558674588662,-1.365817207877688,-0.2309658948457535,0.1416986830427218,-0.1243601066612599,-0.09925705149234185,-0.1444510017139904,0.712751257241566,0.8491547462598779,1.329082151715971,-0.2208707924794795,-0.04958450688867008,-0.5253200172387803,-0.1103148304334034,0.7774212999064718,0.5734073974318835,-0.3277280790790201,-0.877348661802048,-0.2536339967429485,0.4865034786419442,-0.8043677574024567,-0.2283039667212974,0.1306650055515397,-0.4022822925565847,0.2160595201923959,-0.9447754279632052,-0.4936415933410369,-0.5500433815501155,-0.3446349320712495,0.1261505327894528,-0.6954803229622994,-1.121403737307143,0.7796577437303696,0.4481201657642303,-0.4095676834257732,-0.3503113226145774,0.3094046122960287,0.4429035254298285,-0.1933095550814102,-0.1977723269394153,-1.020423967274278,0.2820600627235074,-0.402594068501812,-0.08130420571181651,-0.3860327677159138,0.0003033578970202882,-0.3580714446365993,0.2012847397521443,-0.1273060137789953,-0.4760005105336358,0.09772149106248273,-0.6935303935066129,-0.387017679097558,3.048221437402883,2.626071976715691,-0.4477320334086844,-0.1790722609667652,0.1139950158061952,0.5149429379814936,-0.1201270941063643,-0.3226492522704488,-0.3217764766293102,0.1348068126954102,-0.007130359660251672,-0.1339749364002527,0.05779721726615294,-0.3821987231726319,0.008474843102275221,0.01912242735385955,-0.07172280456879247,0.01081785799966907,2.648636680726804,-0.1746229075166487,-0.6281108123119677,-0.1998692906033909,-0.1972304744965801,0.4826496233141326,-0.3776328891787989,-0.186969480446619,-0.4709990529402114,0.2170718839316648,0.142885751798271,-0.03876921993828597,0.06104197101545645,-0.203263532524151,-0.162935976921619,-0.385649302409193,-0.5260600116244847,-0.07732547640413025,-0.180106824229215,-0.260833391561803,0.2849377374961936,-0.2101589092551198,0.4502404032057638,-0.4906982944869321,-0.4496942950231997,-0.759939431009469,0.6465465758738875,0.03258372590323362,-0.03678393928457387,-0.3982819506955462,-0.2905650277298235,-0.2176485013452216,0.1683789128981075,-0.1733345127363411,-0.4278665430255628,-0.1678602686608875,-0.1940832874785428,0.7297081854453746,-0.679263168425877,0.2128501382091389,0.3057510109944718,-0.336760878714064,-0.4778468141716066,-0.02402733514275179,0.9981918450288524,-0.2812562201098427,0.3297698402265131,0.5909124100418151,-0.9623310580819388,1.038717146686405,0.1583722101820896,-0.1093316564371467,0.4110208229633881,-0.2529844859722357,-1.086890940793473,-0.5763632865013463,-0.884938644031397,-0.08601743617184847,-0.2875772829472465,-0.2420312239111471,0.03853740388414591,0.09575199126018846,0.1100121742806072,0.0251460963426124,-0.3248427008904761,-0.8958207265301977,0.02310513505905757,0.6650329652006235,0.1712268772902283,0.1505246117346809,-0.6539777529047378,-0.4883295686218456,-0.2839407919240399,0.8562407826980067,0.4486978322463815,-0.08074042279903683,-0.4185856666710355,-0.04697009812761471,0.6159785753850227,-0.1712344837811426,0.4768044101180288,-0.1197747208896575,-0.163440110746511,-0.5280397528990856,0.3613708371032338,0.1806189653140838,-0.3283878876069744,-0.05692119976970427,0.3158020248984393,1.210376137434912,-0.03841628480293659,0.4488843660843066,-0.003340013747546561,0.6354169625528172,0.5363721668179212,-0.3386136103529879,-0.1288883712152513,1.4943035311907,-0.2831246793564156,-0.2557341516937341,0.3564407548976697,-1.527497820788603,-1.001905493832368,0.6963473548675834,-0.3508227407720403,-0.4659375073452373,-0.648121065886159,0.2359652028927688,0.4847389962527406,-0.2328462924470049,-0.02695441093644783,0.2761428003720915,0.2233860808440319,-0.02480752117720721,-0.1176399857815957,-0.222557707348787,0.2391396780441475,0.2687296376805888,-0.2645570338753283,0.06005153638862053,0.3663216870608706,0.4125104393980163,0.1836809135441219,-0.2512527219752008,-0.3386374427583218,0.4975535838148517,-0.07964803403754245,0.06893158077427242,-0.7909707590781707,-0.9606732014610693,-1.236654039064432,-0.2327185718002534,-0.2618374363114587,1.273615112333596,-0.7370356076519646,0.1761358804340307,-0.10507828792755,-0.880392311381842,-0.9648844565423409,0.7769498132278717,0.2935644139380053,-0.5059401099304973,0.06026200702388011,-0.3574600869737351,-0.07127670951680802,-0.1007311061133258,-0.2697197276636042,0.1675280005646254,-0.3838403087604405,-0.7151081559550393,0.711951726319476,-0.985488591760208,-0.2608001471321333,0.2576914971683147,-0.09707748053241154,0.1316545069324638,-0.1153404469348285,-0.3927765743058035,-0.347484891325618,-0.5814813805675774,-0.06468233374257999,0.09069454402519384,0.0930141211387438,-0.1637988883506051,-0.4475351816425406,0.5094343323284503,-0.01049410300125776,-0.8835962989750359,-0.1117003649161828,-0.2045752481764338,-0.231251051007855,-0.006792468140295307,-0.1323060091003972,-0.4285493487953474,-0.7288833756067141,-0.2848493085538857,0.7098713321698488,-0.8317420169821035,-0.11214845591733,0.5789362758254321,-0.3376504213099671,0.7381736033705921,-0.3437096045758078,-0.5577979438352771,-1.430193139293007,0.2183226718451015,0.1218674402607935,-0.805308912784516,0.2901890958015195,0.3021855726385083,-0.06103167584684791,-1.068680434248008,-0.5830166901964536,0.07581218873475622,-1.024357880155156,-0.2183402931976216,-0.0175401123200286,-0.3060113743378108,-0.1386489234034401,-0.9650873449933772,-0.2391659765206657,-0.4980236410733827,-0.5699311105425021,-0.3709671409195055,-0.02966355247893353,-0.4927353824825467,0.2017532581821275,0.6901256243580119,0.1532938929194793,0.07109998669191807,-0.3031678476453679,0.1525722268206569,-0.6100765944796076,-1.330321453897926,-0.4242949860698806,-0.2467922145841145,-0.05798833499787451,-0.210038110687324,-1.324641056754316,0.09499633499462001,-1.291169293550855,0.4015175124986767,0.1135604916825363,0.7074224291880578,-0.05140073543248547,0.05258185994839164,0.4894984384957269,-0.3189738909886019,-0.6454658984796737,-0.2940478375637468,0.1707493291324358,0.2364343348784466,0.08849038569865085,-0.3458546586018976,0.4247582270005021,0.06607992514192465,-0.2847502637421007,-0.5814191553902193,-0.4254579823601274,0.359206062141078,1.068314724794611,0.1242150605493272,-0.233588558930934,0.3630525551822375,-0.204305903227601,-0.0562181162185876,0.2663597144004771,0.2907569225945704,-0.2665773330981685,-0.6510728263202571,-0.2185055318468855,2.387807254624546,0.1005208219416816,-0.459477441850681,0.4573981210524426,0.5558957613941813,0.1677155926543487,-0.5482004750444068,-0.2717753318783671,-0.02127321554477791,-0.05401206169054129,0.4800375276599795,-0.4407090588751615,-0.2057729742532725,-0.1592636453594356,-0.3634625826488557,0.271303017286899,0.5687715896812229,-0.3849492847703928,-0.1782615930783751,-0.2234338008721346,-0.098315378313362,0.07877529211970187,-0.3425955631026371,-1.249424308974516,-0.5266284706110058,0.002338316033940491,-0.3663405008620239,-0.1623231969255555,1.627844274953004,0.2009383887125095,-0.3120660487006916,0.07310997830984915,0.657604548094849,0.2948966039830867,-0.3365547671215538,1.271351288168039,-0.7388482414396162,-1.025933456171531,0.2704672861336242,-0.08192596238115664,-0.1594617958593505,-0.1309613910641322,-0.1772876342540369,0.06164065383671804,-0.8242865636703148,0.6674840870043652,-1.085639163851708,-1.076480193422543,-0.9663218486787826,-0.4038397163460703,1.290961910328114,0.2672132356327031,-0.6241047026407383,-0.301949617665829,-0.1918589212187863,-0.1648238359240639,-0.5161275852091679,-0.02578251838970976,-0.2583650584731427,-0.1681526194633812,-1.379166593402046,-1.046502301343083,-1.01693268547285,0.2759737898476109,-0.2756498148720002,-0.5848034808892609,1.474970943340046,0.3293268768178239,-0.8251497369347648,-0.05872913105724897,-0.1777763386876978,0.543136063131948,-0.8956708898015453,-0.9616586620705074,-0.3594665595309972,-0.1042504528840696,0.07358713160402502,0.3085832375231339,0.3078542605913779,-0.273182607986995,0.480185914928664,0.1392668018753425,-0.3147255818346957,0.7048514634747225,-0.5366095896003665,-0.04631688114327111,-0.5612876566610688,0.3058672626827029,-0.5134800849704036,-0.1506525601925383,0.0916229885038279,0.06970968968224805,-1.060932706293142,0.9399198524859003,-1.06202682270658,-0.8215885241682687,0.1524574463829116,-0.1653498498982051,-0.1980846161013425,-0.4125269696702563,-0.4738908010703724,-0.1342369566419573,-0.285236544591807,-1.361711102611006,-0.0656726939597299,-0.05631746685256105,-0.02118668371499601,-0.1932274147381993,-0.4761544303112261,-0.3245217486411172,0.4843007461975215,-0.222562835245023,-1.028511415533,-0.4772788849418125,-0.8266811990018408,-0.2755828782930194,-0.2681053349946692,-0.1561199558961215,1.317783361386127,0.4409952289981379,-0.6072552918880403,0.2081392964174775,-0.6126718248724092,0.06027053318454275,-0.9365159611662,-0.06460288753054783,-0.7464332180237124,-0.6266958041191439,-0.409131400703086,0.07670987353827301,-0.4701962914050819,-0.2082025128095024,-0.1761848745317731,-0.6342515433357478,-0.6933663987292557,-0.7382325372870281,0.2492502957390655,0.169874697300867,-0.1920488911683237,-0.03889699444709203,-0.1585646468825736,0.1928228415619656,-0.482482226862111,-0.6253318862877207,-0.4833958062507088,0.07943902429730011,-0.4133393523708093,-0.2626689647077511,0.4386333183832036,-0.03785382161424405,-0.637590415186351,-0.7062655922602927,0.03330663136422025,1.443702073536575,0.8161049289198815,0.009387989195807715,-0.7169785348341604,0.06489802734493677,-0.1898288984625365,-0.4975604724121533,-0.380926979878789,1.597464692487155,0.8229672559826431,-1.167912097541659,0.3608056486980739,-0.3992951901185524,-0.3321243236263549,-0.5537583364217613,-0.7297733135314283,-0.3914110332588569,1.536762927008401,0.5370160949351912,0.3900896922754263,-0.2416474478953911,-0.2736019613696445,0.2524743016828745,-0.2438821860123511,-0.1161696314322217,-0.2664521015533546,0.3012830978750934,-0.1043851414573038,1.063319202592108,-0.003231446367791033,-0.1366834758896596,0.02186784910933239,-0.1766891076194469,-1.236588485493776,-0.4948606919844704,-0.8097790083143593,0.06075912183830763,2.758199438732606,-0.3667600257166987,0.6704100720248494,-0.5877774034787984,-0.2727969571395512,0.1261554253800055,-0.2708229087021357,-0.01678731161520318,-0.1913966404240987,-0.2943510056331757,0.1011205261357098,0.1786805816515849,0.1464800750661716,0.8318054756709254,0.1237964469238586,-0.2789642418346953,0.07633619513900368,-1.148144093768352,0.3971752823545013,-0.3843523869419347,-1.399078431351617,-0.3266244206275427,-0.7665184132055202,-0.2987957485762099,-0.3282856898541097,-0.05790528966656411,-0.5567636888910376,1.627900566982039,0.5397628361103044,0.1543025375891659,-0.9257977080981451,-0.2460223885924479,-0.192357288818345,-1.371217949688539,-0.1815418232604284,-1.390768791928137,-0.2712121568609068,0.1099165080672855,-0.5629042930028705,-0.1930778485692772,-0.05337802400301429,-0.08928300113445602,-0.310927749242864,-0.2635188692537376,0.1081914870227311,2.377104292905799,0.0420299316971384,0.4361914351824078,-0.2623789599693575,-0.2482450219829337,-0.02624143300293922,-0.3054729964971106,-0.01833195574281144,-0.2845744523290679,-0.27819227893965,0.1309369172347147,0.5546119036519608,0.2601636568175699,0.02118142320406404,0.378022464813977,0.5286482146341279,0.6949118902050549,-0.5465138351541493,-0.2511584725853503,-0.2684490262368401,-0.01574633438226497,0.4110261182255125,-0.2649618951817251,-0.2414925959483539,-0.2834003232078529,-0.7141068533662597,-0.6275017897282159,-0.2101175584472508,-0.4589814259239275,-0.2627958058302608,0.7207450728947988,-0.706252518128639,0.2167446125595638,-0.1914746833479586,-0.1785059990263314,-0.1365357789381858,0.3104396257358725,-0.09163783165086749,-0.2213177317693668,-0.1690907701684308,-0.2440702460062857,-0.1624859342197676,0.3842951111895158,0.0110297086226739,-0.348039764218325,-0.1869997286712554,0.1942186356006466,-0.138386895017784,0.2083490481734515,-0.1220634821659705,0.4357011313717178,-0.1631034676444946,-0.005095009813378292,0.704817853015126,0.9960094888775426,0.01742079592230908,-0.2154416094741576,-0.1525549629324363,-0.3713118476600316,1.631726432612615,0.4832120546983246,-0.06480314776127188,-0.2528571546454313,0.2157764902907707,-0.8945020372924155,-0.2444168346438496,-0.01333816857196994,-0.4842821281954311,-0.4705895121094318,0.5088420931778763,-0.3307995224825795,-1.800369146968068,-0.4840349748229094,-0.03354749974618898,0.3173356925224527,-0.08392669494773045,-0.1249740714670048,-0.0567051660926722,-0.1117252914646607,-0.7038387578679336,-0.5649739891617404,-0.8476577069153377,-0.5654092216848994,-0.8441343469351293,-0.6242257732145212,-0.3462473015534382,-0.7983953131439254,0.2050149603549851,-0.4883949759240984,0.1370483460779482,0.1629214417706125,-0.4685831908322079,-1.185598729967509,-0.6231787575162994,-0.5224873767685034,-0.318554629287187,-0.0928365524994494,-0.3508309863334835,-0.4351662577784965,-0.3565214786397178,0.250352434051566,-0.4712218561084767,-0.4852906077571152,-0.6132812113468253,1.144754047678701,-0.1172310637812033,-0.08593271088528645,-0.06829245979200303,-0.1552134526074566,-0.4203033332099896,-0.7066709029094985,0.3455705080661341,-0.4211940347159924,-0.143338490828522,-0.3235631769793937,-1.079397312029294,-0.1185476250401349,-0.6733513463384628,-0.3928689818637826,0.2719338777701804,-0.001409725582065312,0.4612708884166504,0.03495001411323206,1.573348467586013,-0.2104393581765855,0.3354777413864166,-0.1373423570897971,0.6931319780678111,-0.07068129646317779,0.1015744543696922,-0.04389448712680005,0.456432379857079,-0.1718868723961971,0.1686303478050591,-0.4976808719156355,-0.007813846936696378,-0.0723386094372923,1.344660995786723,-0.3448266900816554,0.08918167952149691,0.7741642924957294,0.01746032899313004,0.2649950605003318,0.2413623731174924,-0.1161687700721443,-0.3183916420681521,0.05660995676221941,0.2712950434123639,0.1489568389481615,0.6008859177963756,-0.7890542072505753,-0.2434962200781813,-0.07916189684647069,0.1275849829249465,-0.6924376199767004,0.0218678465003007,-0.1542644528286335,-0.1387064422587364,-0.008459807463366751,0.05067821356836461,-0.4583012571612236,-0.332087751837233,-0.3076164157131481,-0.06382532459978815,0.4168064211560797,-1.30563804162363,0.2831117141513856,0.5393918625273517,-0.5008993650699929,-0.2489706192127088,0.4929715748746836,0.3692174717554043,-0.07199330266525281,1.664796945040394,-0.1384346809488896,0.6602384077858103,0.707104340823114,-0.841124956643802,-0.2967273628635659,0.5977456133868436,-0.03408976888462398,-0.5632809087066031,-0.1796187437608155,0.1034840915909037,-0.3293281584377003,-0.4813743258216458,-0.05718046909063932,0.9218781519382895,0.3693884332422424,-0.2522781620264639,-0.001967461498834029,-0.1561225147136135,0.5474300774526941,0.2346656556919053,0.7530108784153717,0.2774579517672802,-0.08320569079473837,0.2649232583569149,-0.09484799460153746,-0.4655231193402617,1.85921429868107,1.565682010914859,-0.2695429388298972,0.2847148871471619,0.1090039256402442,-1.264465703809271,-0.2208257483650271,-0.1423081763062912,-0.3335159976039589,0.1970094718214854,-1.055544562810623,-0.06709238939639617,-0.6190549103102739,-0.3155919699171398,0.08386370458326271,0.009521653894796256,1.38628573665886,2.827938858657141,-0.2024693084455974,-0.1116176770436644,-0.07788173197044435,1.070580411909529,0.5126530300002298,0.5524079442446745,-0.3155155226898391,-0.1574433542258298,-0.06704721032059728,0.05259372400211811,-0.1282012896850966,0.1096724257147744,-0.04778343767966808,-0.175273215475721,-0.06878881304324003,-0.3583011697562815,0.1299499368531276,-0.5150322780417279,-0.3465769614987478,0.265566723083239,-0.2154753496249984,-0.5988626365219126,-0.01079154746617171,-0.2273016335922863,-0.5588346447648608,-0.5224780783722951,-0.3974187480515119,0.4134210119061008,-0.9077025380661933,-0.4894987797138536,-0.1219940773139608,0.1535166726532773,0.1505614924720864,-1.074972065587931,0.3962165643029812,-0.4680759051230563,0.3566634298937277,-0.1201396695884598,-0.7753963966727091,-0.02419680446624911,-0.6475581152407824,2.045532354895871,-0.767028369608909,-0.0850075199847234,-0.6699397734402091,-0.07244609510900994,1.221483226638766,-0.6730670386581161,-0.3763626888696492,-0.02308537182069673,-0.6795946190430385,0.03265460805880431,0.001095402145766928,-0.5540353363773902,-0.3533611481837896,0.2948109927511862,-0.4774574532002006,-0.1362823510994851,-0.2314149527372003,0.04977942263123979,0.03715271029020153,-0.3509054235229831,0.0508763750202556,-0.2562864836574088,0.004931492367425046,0.3240011217720425,0.1692628410406691,-0.1001114470707466,0.4382202859260287,-1.161914416813014,-0.5702159255006938,-0.3529047556592689,-0.08371080000326768,-0.1634354583526743,-0.3334395514047084,0.2110635019543569,-0.846371335115371,0.100614918553274,-0.3089440023878385,0.3381000810066428,-0.02196548807834996,0.0450428689043197,-0.2323776615284869,-0.08207298819155572,-0.8267774513273461,2.230116472192706,1.315973733117142,-0.3939532182603545,0.5942057471381651,-0.5290267571098621,-0.001775229428551961,-0.2214292035604258,0.2153420166656849,-0.01699791956287419,-0.2164390618587386,0.121223490630302,0.3050530828634746,0.1965214931204413,-0.2841466807184274,-0.226717044048792,0.6398587202143502,0.4506797329129853,-1.138774142581413,-0.6823510939234914,0.3289080617260274,0.06842635517584987,-0.03557350908062337,-0.905844833052403,0.06758846659346714,-1.322212761793091,-1.061728946936568,-0.2597665615353644,0.0363426110477015,-0.2054251405732526,-1.020807152330871,0.7779829054864995,-0.08199756927913339,1.289578960319108,-0.05671003310137689,0.1735032443223457,-0.6863558857939877,-0.0620708357145342,0.400429790179526,-0.1502988109821751,-0.3235700323289997,-0.0353998381277115,-0.9823809459838581,0.259249134539611,0.7839479675526793,0.5676731377795389,-0.6805772374833119,0.08008216405511435,-0.3248043482146878,0.2097609042686668,-0.0799409177617974,-0.2939236133077252,-0.9604434077735383,-0.04167723501549424,0.3634856412874388,-0.2405019313437967,-0.4624322894698936,-0.1353667079954985,2.040734644061128,-0.4823993224418063,-0.2156103713643424,0.562034922557001,0.0470859238565681,-0.3286304336309677,-0.4097278540311976,0.2347746638695407,-0.01052271951073834,-0.2306291109870116,-0.3714794055422419,0.2360781592354351,0.1782985493302147,0.06588717763293037,-1.004179130004935,1.206759164458843,-0.0396363496239936,-0.1175737131622086,-0.8863492957857901,-0.3675048141724982,0.3446046734260377,-0.3414809360139389,-0.1333535111592309,0.5047742401190586,-0.02241524107731704,0.0192629914060881,-0.1870254644084453,-0.2716165040222092,-1.043634109189571,0.09739475370597719,0.0980448300512968,-0.8509815919169906,-0.00798860659013001,-0.3058881434440846,-0.7356335379155426,-0.3498346291092149,-0.05681099075604169,0.001088859481232234,0.660971176775737,-1.208134102827353,-0.683767391468184,-0.9219380946640117,-0.1327257560158462,0.04104403382980589,-0.186855195782744,-0.03899353423603296,-0.3381650396703398,-0.6398524876019639,-0.07835132563006504,1.069840039050633,0.02555600894085173,-0.5354721249085771,0.1075213626990478,-0.6616390050272853,-0.1764999459647315,0.01637160940955823,-0.4770630603075759,0.07721402340493456,0.6920400003648767,-0.1847530731289924,-0.3300582253180197,0.5525324029970151,-0.2711980508035409,0.7365201567770093,-0.2472944654956583,0.06774155457109454,-0.1018944750479181,-0.0421475193230046,-0.5179094319258363,-0.1263037295984448,0.6966432962666613,-0.3340323806022508,-0.2412849450512582,-0.0238836772060419,-0.9457031775684677,-0.149107087664434,-0.7973112196809624,-0.6401693661844881,0.3124033112292028,-0.7675295726229709,-0.01443978224110983,-0.2319236868639494,-0.2779225516961095,-0.009272412907044962,-0.6674123613877452,-0.4449467916068277,1.046823356827613,-0.3523997769603348,-0.01923134691893086,-1.422734655557771,0.3014942321516584,-0.7113053013472574,0.1599085550674387,-0.02517062103030827,-0.3896416133445342,0.3949441573409816,-0.3315629316819756,-0.05554516260842531,-0.2102825051353956,0.03455825088819623,0.2070893160002997,-0.274330855984811,-0.1070140215801588,0.1871409190469903,0.7365795046281131,0.1465469405004412,-0.1075258295238583,-0.2593985673247717,-0.3384911281944015,-0.3831895553315576,-0.03337036604252203,1.235142357012468,-0.1099220178587955,0.1327209822416073,-0.216337789687786,-0.1269819514685532,-0.4342868210884115,-0.6350097100338492,-0.2036130418712899,-0.3137867217698226,-0.02058946679631946,-0.585963830907635,3.576650712166658,4.187592809026971,-0.2264677246956894,-0.2730615593396309,0.8560855515674189,-0.3970222176514216,-1.312192112972662,-0.5792029969831949,0.5417360266404868,0.09485603068288019,-0.1098289821986523,1.40725212524601,-0.570064479373561,-0.01059414693431667,0.1392274205322056,-0.368144331675011,-0.06810111287564588,0.4678929512782247,-1.048255673420367,-0.4334548017798186,-1.058469741193556,0.7092911389526692,-0.1020399475785154,-0.04807709358842158,0.08748339494940965,0.02790008687496415,-0.3370437864951607,-0.4833213566580314,-0.03482529922988127,-0.1804068068883331,-0.4853858088741118,-0.5587394092981003,2.412834529705159,0.01845891020292525,-0.1117115821068803,-0.8620627075523128,-1.048190169693271,-0.2643258058641565,0.4336024601675275,-1.471987201575266,1.828224480830955,0.0898216959957779,-0.1451977006256653,-0.185686004193183,-0.9672928646095806,-0.6628343785248498,0.01275843353999953,0.4103976004381187,-0.08093131447535838,0.08789949727198408,-0.0496476915147256,-0.2959166618005969,-0.4864443139045223,0.2804908728294333,-0.5861170604586937,-0.530358235835068,-0.04053514941277361,-0.4313047830901717,0.1588272929425141,0.1588807788313933,0.6802653255388237,-0.4493278074966903,0.7581163902564054,0.09464149810274745,1.367463040827348,-0.1114171873630181,-0.001959594337380563,0.1398359738493846,0.04777737506806657,-1.141829072659149,-0.1561831279014379,0.09419986819380455,-0.4705347819662363,-0.007938217813383391,0.9979617539103692,-1.112791365153301,0.07626707822201746,-0.1641969790153678,-0.3698755408077248,-0.134243104975298,0.1247336258183525,0.8609362169901301,0.2095845829799906,-0.3774380977807407,-0.3733689359252048,-0.5923734921590796,0.04056218225104922,0.5945096984335954,-0.2623717897509716,-0.4691157159846834,-0.3133766734339117,-0.5547674661156354,-0.173862736868194,0.4321106140951858,-0.3913097373660233,0.07585003568798963,-0.3500628658807829,0.594609922067845,-0.3064860843452785,-0.2966587509013405,0.261332554056205,-0.06209524667909155,-0.680354959471096,0.2062708483190139,-0.4443873553071474,0.5924191912871293,0.7719657619529867,-0.08275573911124383,0.2176131804500989,-0.2231640617567248,-0.3913634392700588,-0.2087998586085764,-0.1284206396209019,0.5166271409986323,-0.3089484906519348,-0.1108995106471464,-0.768011579191466,-0.1936866609745757,-0.1037722776901451,-1.310975059959402,-0.1738083393440826,-0.2081642597528636,0.8726092771184122,-0.4669281767256179,-0.1418226343751598,0.02453755152241679,0.1335104141859157,0.2941557003780679,0.0006946625142428407,-0.03719297217834605,-0.130289810766075,0.2695009808699351,-0.4772181378243202,0.6981714164135782,0.06498887579604606,-0.265668608377984,1.142979812527159,-0.07702381499425426,0.1112910716907132,-0.7226983592750975,0.308241874474439,-0.4151791205419194,0.4225305396098614,0.932090808647562,0.02414028928863557,-0.2768489456561633,0.04630875004383974,-0.8138019857575028,-0.3090415099669491,0.574697473350153,0.04831694022744908,-0.05196235528220298,-0.6183034560483942,-0.09102297525176201,0.1879905716895291,0.5235384475166983,-0.07402591949249374,-0.1718878215513337,0.04645097927079876,2.83449439232272,0.4663624676643922,-0.4352215749760573,-0.8383635183685157,-0.6208893885306015,-0.2201804850322303,-0.553453452078864,0.2270562454615988,-0.2723612904835497,0.1801093977926388,-0.3243077046668366,-0.239294349346298,-0.5590103488819551,0.000776764556475552,0.05275143757229503,2.089699942831322,1.643769197787995,0.02052712188707444,0.4473428838076469,1.699838188933056,-0.5857597752859881,-0.01623717087899305,0.04923363283170566,0.3777600487605505,0.04577817187308205,-0.9967025840256043,-0.03037170072596474,0.04190029862678401,-0.6229694479455994,1.124534077803622,0.1556563334503878,-0.1727932824870622,-0.09166150839378821,0.3629597589427882,0.8294912989089306,-0.002242640026191957,-0.4918900100836309,-0.02285905517918107,-1.400431815950907,-0.3680140576808152,-0.444835052257776,-0.2227059916872375,-0.4221954854337453,-1.06740987356637,-0.5298951374661274,0.4259433077438463,-0.289538505233041,0.8278733173558661,-0.5363094961725728,-0.3250672531788411,-0.3354993523102929,0.6351249220761043,-0.08872428130067626,-0.4749609752650605,0.02607025270439704,1.512420564783956,0.6028088655048571,-0.01758173294534748,0.6253349307187972,0.2924610480523835,-0.1681796755806298,0.1735741244970047,-0.2147856854279407,-0.1622034834295064,-0.8214955337791491,-0.6717525772868378,0.03909991239421583,-0.7304490842786412,-0.6527441940089885,-1.346402486547397,0.4226049556621634,-0.3878661373759761,-0.3389163150655796,-0.7585594798425868,-0.1362645557138898,-0.1492452251088458,-0.02623664749215397,-0.01908408653912634,-0.4673455683488876,-0.3878689727976887,-0.0696529694185013,-0.0003830481983088172,-0.2817489118528496,-0.4550129887882306,0.213996375596387,-0.041690686082133,0.1145504953123315,-0.451492044197277,-0.2298284865725282,1.404742026896787,-0.4073033660747213,1.581530593156477,-0.4881698845616911,-0.5148964270617991,0.02937838978521003,-0.221633785965053,0.2910763902915992,0.2953677523977367,0.04559168314228701,-0.3637053720810624,0.05206957022761077,1.643046326088849,-1.047145816505436,-0.9122999467439701,-0.1061400767507627,-1.155744775973636,-0.6285237655251832,-0.3791092841334125,1.527767779307046,0.1129070929900629,0.5427294424186233,1.557216650661213,0.5870979362736238,-0.3973539372682836,-0.9872377990689029,-0.001063340005738653,1.267159074842048,0.108318825444169,0.002030854223567811,-0.03393927652284216,0.3115731272583349,0.05669140965619341,-0.07622678280272296,-0.0554472256282719,-0.7373535833793522,-0.2949452564004467,0.293913783119643,-0.08879478417708063,0.1619656254218365,-0.543084451721207,-0.5076936883065649,-0.5149804412182749,-0.7732855171792663,-1.063808677268012,0.7097598858424895,-0.233410688616096,-0.1449345180701796,-0.3770382356301437,0.915776944078861,-0.1840168810371032,0.5740567772103081,0.3560584847405789,0.02320752289895394,0.7635831056971542,-0.3141842559693122,-0.1843536457082852,-0.3406138965917463,-0.6203681049375678,-0.6009114116711474,0.1263959061552863,0.4827725552471869,0.01711982078258015,0.710237488898279,0.1663452289930034,0.01382427624538735,0.1138684186117874,0.5410359427262581,0.6463554519182892,0.7608533043223841,-0.486995896861476,0.150431041101195,-0.1541475323744045,0.4899245402159086,0.3748224729136855,0.3155815623639704,0.0937562403124497,0.1092422237983287,0.793796925509936,-0.4853026129561364,-0.3307900245294854,0.4639566599436504,-0.3038218968155804,-0.2626518491041221,0.3790938530919782,-0.343009511376396,-0.1212634468039063,0.6446317146055829,-0.6978048748481396,-0.2625663879350465,-0.6483062858079498,-0.3564951413546568,-0.3523839875388251,0.1553281717315859,0.1082225173288922,-0.7528736286476099,1.444223271385992,2.138651777870126,-0.01506540446431668,0.6979123360590825,-0.2386677140401095,1.824431048648035,0.9264745447309077,1.983575165924542,0.7421823758655695,0.3393083726390067,-0.1850318282657217,-0.2935752526635583,-0.5586742627208691,-0.1274733043336661,0.1602332024358595,-0.125701436356182,-0.4827525697414289,0.2628144877254994,-0.05646414752887997,-0.2062091229978355,0.8931753342435129,-0.09661182004535064,0.007741367452494144,-0.04892921541703633,0.09334971188992593,-0.5564178877713389,0.09598483685607116,0.1341757908366226,-0.5047999831610496,-0.08659069592764881,-0.004698463559311038,-0.6844687696746349,0.3546771982683642,0.1920885855619972,-0.3978167954388354,-0.8053666553985699,0.3125487055562953,0.002949770148290309,0.4727457479594149,0.1331223475391979,-0.4440856468322965,0.09338294888829385,-0.4796380374717724,0.1006701284404813,-0.2689528728067442,-0.06901856330201883,-0.4685704973855416,-0.1593325163960963,0.1320910146149746,-0.4368105506813977,0.3839015500340656,0.7785943037147709,0.004840661200149669,-0.3319533283944831,-0.2008910474328809,1.379756787658467,1.464716071261402,-0.1647247462429855,0.2152645531642484,0.007355973477734203,0.3987013036954284,0.03886274458256548,-0.8415198421853304,0.9601268926030054,-0.4554531456785811,-0.02194667348622879,0.4173382335523348,0.364252104517465,0.3420906194261078,-1.280464589924436,-0.3305768826296581,0.07021283796764882,1.900497889370001,-0.5551749606708078,-0.0415872475927596,-0.5487042139746343,-1.343291680925875,-0.3553183813220749,-0.3072306810454914,-0.1787502040374257,-0.7877816597101802,-0.8492721753387336,-0.1218844494693717,0.7177389018632265,-0.3255857713447096,-1.051533267411142,0.2266463452988334,-0.4470197611004839,1.307610175493801,0.05627523221589165,-0.15476604823086,-0.4608622915387141,-0.4386385224469983,-0.1467972038359378,-0.05935572470672899,0.07145342683444969,0.4902466331624161,0.07388895268355811,0.5186282708218941,0.2386757064948073,-0.2021237981616536,0.4043502529397275,0.4265559415189301,-0.4577946630603101,-0.1392373430402963,-0.4049691409481698,-0.1775604579201031,-0.313433826886661,-0.1766403498531728,0.04044129789832753,0.2562153552218849,0.8153097137234484,-0.7164117126064836,-0.1022601650326257,-0.1241778466327031,-0.3125941304970711,-0.7389564233626724,-0.1523478307332479,-0.2888598327414169,-0.8585846818385613,-0.4692689854322205,-0.4636430113589791,1.325439431627654,-0.2549546848852596,0.09548220853576342,0.04661103290621434,-0.1564605604078221,0.1281530388802783,-0.4372932057930594,-0.2072149582436766,-0.7912059815358017,0.4845769596018975,-0.04744062913865348,-0.7821139725924974,-1.437597186405453,0.8230720798371253,0.08457567518062745,-0.3755438307355102,0.3994780992572704,0.04898648227667454,0.09866275744903052,-0.2837910922985793,-0.4753522895146045,-0.7519980158628921,0.05037266914797334,0.02839698355314101,0.3340942713812766,-0.0440481772078996,-0.08185644129849445,-0.1668126160919137,-0.6591967286696888,0.4999934245151063,0.483217797978893,-0.2986447987495063,-1.154977403671586,-0.4689842415145751,-0.2948495163124882,-0.9037044000995247,0.6180839290444168,0.03565960698753355,-0.2836142212779966,-0.5960550205685066,0.2884387959534506,1.582356691636805,0.04067220547676027,-0.2760722668686837,-0.508796848251073,0.05001186507368636,0.1496602869681384,-0.6269369385436342,0.4205867660748878,-0.5780418896183697,-0.03909040647771999,-0.7900589955796112,-0.3485238550828583,0.5675532018512199,-0.2798062388016573,1.441386348389974,-0.3123704443172792,0.02050393025409526,0.9947314499760834,0.8370315121980577,-0.4051279482747625,-0.8011077906304396,-1.087752387386794,0.7818948417426063,0.08714923298119494,-0.1128940372117539,-0.4153586951563096,-1.645044471564338,0.8939956661152104,-0.02272168937793187,-0.1672122345250849,0.2712394213926169,-0.1967210726391708,-0.1557074117276534,0.6384576172448707,-0.1742083279362062,0.06660194025700385,-0.09996880543568042,0.139851594228391,-0.02660809048599929,-0.2726284143432852,-0.2180255031683639,0.322274215573165,0.2582302886927736,0.4161766468439792,0.1339942806368026,0.5866375619272818,-0.1189905291061775,1.820671042682689,-0.6632044310065557,-0.7405061515500369,-0.05056225762847853,-0.7190735796007297,-0.09791681626402245,0.5728559474700965,0.2019609625541655,0.0601479206047186,-0.1486935475714014,-0.182910536744168,-0.4970495347903184,-0.09653507479922172,0.981976822845783,-0.1650391270759248,-0.08783340563754666,-0.4151118998136647,0.06102740602115186,1.013509739416535,0.2606527558531252,0.3234441478675266,0.06288728924035772,1.046808778622127,-0.1347436173072036,0.2713034423690803,-0.3234868061559176,0.006779105838567517,0.4300943373351199,0.3322286216536508,-0.007941000021627563,0.166531400615719,0.2097548191601932,0.2530978101262923,0.4904526617788358,0.07803181984476056,-0.1270513130319764,-0.3013823404366489,-0.3667538793328886,0.05844975645857928,-0.6867739354228023,0.8640213945316664,-0.1056947147937473,-0.06556517493367855,0.1235986769150008,-0.6609300540400125,-0.3013897704496273,0.130829554734329,-0.3763130705748987,-0.8319810448840326,-1.306772671185921,0.09255397300164241,-0.4030289839447982,-0.3829764658333851,-0.8664824825587827,0.2384094601165533,-0.01996398891989325,-0.03090692611710781,-0.4162660794969742,-0.273588530801087,-0.1902030696093178,-0.221069934074841,-0.4989193131587163,0.2929415447445415,-0.06285799746738618,-0.2005323729036175,-0.3733967914811371,0.3384900490267399,-0.1880127964639987,-0.2074256311739112,-0.2124555198292866,0.1820169085446803,0.191053275284964,-0.3450027793327006,0.0371543202304077,-0.6300602809962618,-0.6320422645866617,-0.2289076355823117,-0.722908088535439,-0.4363547189838282,0.3069220770121717,-0.1790956817628069,-0.1231966802508606,-0.5262395510147867,-0.08262989720323373,0.1191962228236235,0.06587465250484086,-0.5731323318076863,-0.3056814124450294,-0.2711243320133871,-0.1089843442955896,-0.149628212477302,-0.2822164095420245,-0.1509934433793946,-0.5888338114805639,-0.06518401577996336,-0.4188302624594955,0.03674095633240312,-0.3305136759945862,-0.5256923182017313,-0.1625682773988288,-0.1818912584703886,1.769435527142553,0.6276046736792994,0.952738427322917,-0.1203540738690947,-0.3646726311082199,1.292758989812748,0.2420918663470265,0.2742715614554604,-1.197377452746785,-0.0423961888448483,-0.1904370016411769,-0.2329654847694233,0.1982735313911466,-0.04570177757349673,-0.2302343534176493,-0.1328068323127646,-0.7603050097910237,-0.4062460833244368,-0.2071507333817961,-0.3109602844517377,-0.06259750175651685,0.5476935842732423,-0.7607719716391673,-0.2691220266077493,1.050200109047101,-0.4148420682537979,-0.1185009598207685,-0.1720816556267353,0.04479389013885501,0.3881491580862,2.839977044645936,0.1573350095568816,-0.1471686815891975,-0.5300148655585638,0.2910818594766304,0.06708444123719154,0.1454791134578514,-0.337968837821824,-0.2369963931328401,-0.4308024588309215,-0.6151047461698952,-0.05805114032828721,0.6788808398863184,-0.2537930026480779,-0.03819881020548214,-0.07464794853611986,-0.5697102366476773,0.5350146034601123,-0.1969235525568505,-0.9873793842797818,-0.2156420135759149,0.04761184525179176,0.5749252171346733,-0.8097720978307947,-0.8392312678405035,0.1722376445161577,1.32450734004623,0.1862383207227887,0.3044804446056806,0.1522254563766677,0.3628101750689215,0.2981341338075837,0.2589422630974344,-0.07007989914401214,0.8722203962331102,-0.2200273883607576,-0.2136145662054825,0.186883729692715,-0.1491853874225767,0.02646133882459807,0.4013529545542266,0.3360414064487906,-0.2680067476912225,1.383918725136782,-0.5790219683089852,0.1570700311034691,-0.2140027643000555,0.03814352453283999,0.1597427124138391,1.873733976758915,0.2881951730510774,-0.8364339954519566,-0.4444388975687821,-0.6567801427584428,-0.7178648906722297,-0.06921986034458558,-0.6462500833995073,-0.6581451955034741,-0.4710306264245882,-0.6567714766947985,-1.093770468993397,-0.3390291010596149,-0.7506826289333195,-0.3728865537453592,-0.3163848682507785,-0.4908242890864707,0.02324234976477142,-0.09572675034622695,0.2704191465974373,-0.01525068109868589,0.4863280871365308,-0.2109653740295751,0.105955559972788,0.1202503996797497,0.2453658956525558,-0.2173602079347044,0.03492004882348033,-0.2637659940930279,0.0475167612581994,0.7992789156314374,0.7900332524364171,-0.02908234356667241,-0.4901714166659217,-0.4254477056404329,-0.003797433995254956,0.02736523777059108,-0.1580240284246541,-0.1039724940959846,0.9918836280468422,-0.2645398423374875,-0.3926487148801588,-0.2484748629528186,0.4558847349553339,-0.6486833077216871,-0.1504354023161748,-0.008052817563749802,-0.9233282660545427,-0.2453109988266836,-0.3159079654140319,0.4172313852274001,-0.6542605360857439,-0.1053292382939214,0.4862043123967633,0.3699855973950813,-0.2868416820284461,0.3321316916322481,-0.09626085317964574,0.3470243846170416,-0.3709884269004525,-0.6280237229995483,-0.252250683854348,0.1817303009077086,-0.05373585055624339,-0.6097386901818176,-0.5551712832282131,0.6760062376556425,1.064378556049109,-0.2017635793657728,-0.1691489811487285,-0.1944833057643574,-0.1289446666685749,0.6811792274269165,-0.2630835879727056,-0.3294755717343057,0.7114332181369066,0.3193874052811453,-0.1110395237972655,-0.415357928972251,0.6320696028055017,1.140039957639566,0.2843507026876368,-0.2184789622023338,0.2818674539345756,1.678808611359333,-0.6887033569916551,0.6985618578196028,0.196770559964655,-0.2223010493154476,-0.5644091925036712,1.858020836466419,0.9095065695882056,0.6500837194462726,-0.5423971112295972,1.212491616513503,0.4957397216182811,-0.6208137494977296,0.1970843668368026,-0.2799022281075857,0.1110276508293246,-0.1908551723343527,0.8668591312942184,0.1233936422793781,0.2227272561995958,-0.2415241456492953,-0.934597417675658,-0.02372673325747105,0.1774281205534335,-0.8109930072240956,0.231482008167122,-0.7902563866937472,-0.004153643504581473,0.01211818097149675,0.4610869157883266,-0.4663004234313388,-0.3667328497038624,-0.6717491423271127,-0.7790946290498099,0.6674215119165134,-0.6400290703159993,0.08170233899274458,-0.5706950641535359,-0.9020437501714293,-0.05428627749558899,-0.4633131286260453,-0.6455502072311919,0.2470568970145057,-0.4850982690976578,-0.4374175035597139,-0.4727782768868612,-0.43240827447735,0.2482371066116298,-0.2611342969903846,-0.9079993120616309,-0.127115590853375,-0.5865388507179948,0.04721833713925654,0.5608884809264428,-0.04012670897280265,-0.5107803303534908,-0.103487803008896,0.1391681688431882,-0.3732103837510185,-0.5825633695983454,-0.04828444233733023,0.1412316869448524,-0.2363289112262801,0.4076269093652607,0.8465327552653017,-0.1003433690268457,-0.3773182706578296,0.4033530999441315,-0.1984298166992298,0.8286854008352572,-0.1132165179198256,0.2907726138294178,0.03225780300116875,-0.9180138142792673,-2.183621563873907,-1.310075080591281,-0.08777048753150822,-0.6603754907412656,-0.4321030097207681,0.03172123713536952,-0.1673279344662257,0.1708205016041759,-0.3844159747327682,-0.2793851436258369,-0.05883427686872614,-0.5918093667777587,-0.4583891440246352,-0.1555607195427056,-0.8069066976863909,-0.4419917948554529,-0.7259169323815665,-0.2746882886875862,0.1290017040057369,-0.4139886770065844,-0.347025247400773,-0.2393996488879081,-0.1024045408071948,0.1923305298319364,1.562112463949176,0.1642596563871297,0.7555339577372221,0.2148201631357281,-0.2600124547541986,0.5010952483141847,-0.2708835087036774,-0.8419356309514571,0.06788016886050753,-0.2347005402403368,0.02932110044602175,0.09252488784930206,0.0184843034686841,-0.41454127864808,0.850286211004317,-0.5491030983724974,0.6495609150619873,0.3463473456188261,-0.5245390473760941,0.6433426977330418,0.4066998444783741,-0.1978833065487071,-0.5268228179027366,-0.2025662627989214,-0.6035192756656197,-0.2397820758619994,-0.3232032815206567,-0.5828919831496188,0.6464516154571915,-0.6395849201621813,-0.5181381450025274,0.30416562099108,0.0260330201360942,0.3237321461048145,-0.01595399571737256,-0.2830502450147453,0.4309219851945378,-0.5500631602105031,-0.3375049347781351,-0.143447538889141,0.3537734293155677,-0.8815256558624244,0.02011492611030851,1.476030902697798,1.617902293282387,0.5618985040015009,-0.1364032021750525,0.4092927428838859,-0.08983950511924094,0.355032942558146,-0.1042184625975422,0.2901542492541894,0.6325090071461995,0.4765753693521824,-0.2421955826551483,-0.01184020647355074,-0.004506760640238249,-0.07069146783846786,0.2387477519621079,-0.01036785903305394,-0.6435314877826325,0.3328939535842531,0.09467545761228323,-0.3075515505315543,0.2300139803063727,-0.3101920722071632,-0.1251202569898908,-0.4594481152621921,-0.3715647291316179,-0.7654163710842973,-0.07164356766885725,-0.08327891510822244,0.1722972997427631,-0.6626526715400575,0.8260390500050938,-0.04439533967495296,-0.2499576740744953,0.3692681764167808,-0.1489022401395522,-0.6230587869228923,0.2323092361909937,-0.1142450444213551,0.0586726992800527,-0.3293205547078647,-0.1781988735644,-0.5411842071597123,0.3503467662590232,0.6938679086602253,0.2827570995632872,0.2026528316606227,0.5812456939086384,0.2978602949229781,0.07606832267133747,-0.07298902628374836,1.141017572726283,0.08303072512124146,0.06461343114163129,-0.2685771703939334,0.1598800619093616,-0.3423391293035165,-0.259913479913374,1.547317726940554,1.817686594179193,0.03888827738460488,-0.1628517271353032,-0.1783127400585744,0.1538984141761209,-0.06825991419700951,-0.08199234536816667,-0.3730577548588817,-0.05434241444563355,-0.5764151546650675,0.2101262177175802,-0.08875902434872886,-0.2567518799789814,-0.1407889066517003,-0.7603200366196899,0.2271517893170842,0.1331440081903014,-0.6139838229992461,0.07318394985451791,-0.8384206074466225,-0.4942998097791544,0.4601664198267941,-1.103736503932972,0.2812101205027109,0.2258760188684263,-0.5661776475801861,0.1719385839114383,1.887036079247351,0.1243204466447579,-0.3163077210588304,-0.09920206541354418,-0.4791961159651014,0.02366554188279207,-0.2902626452538772,0.3873015570696285,-0.2702202892850945,-0.08265971626624795,0.03676992911643436,-0.06338108539855482,-0.05800906595998397,-0.6117629246877071,0.9029222863507196,0.269383486340756,1.860310693568904,-0.08867744574578947,0.3508542216685764,0.05781814813516453,-0.169662713973628,1.212941177046614,-0.3353015117916026,-0.3277762690753059,-0.05217014420623468,-0.2462444694271443,-0.4141707498500706,-0.5211523832463523,0.08492610637925711,-0.4373039711319209,-0.258434100651153,-0.3698296642026384,0.006637530794457613,0.0001950128252027676,0.6923967800559029,-0.1695718624526025,-0.5379754650462102,-0.2875325548344065,-0.1632594925824089,-0.2082966407527717,-0.1535897230819516,0.06629412899822354,-0.1585619173427424,-0.4421474758706732,0.4171242880217835,0.02044290236027414,-0.4250081501906527,0.1781172178114557,-0.3774351712717255,0.6790638470274796,-0.3691651824692083,0.1558248287947211,-0.09199252172518879,-0.009674800276069703,0.2135386189201415,-0.8764275845428935,-0.1146281797653351,0.5763794832579786,0.06018220247928981,-0.2513522723128935,-0.0740015670673491,0.8823318516466071,0.3273676198452047,-0.07605349083583755,1.306539964588363,0.2812342422991796,0.1320989168271057,0.06122313927894574,0.1069314926080724,-0.1285549456070598,0.06452116744413797,0.319453916561673,-0.01728524647324538,-0.5340086044231082,0.3829868272891166,-0.1152965370968105,-0.1908319012276209,-0.3563249565575499,-0.5986863861156178,-0.5306096046550872,-0.9339951624150133,-0.4490816004099136,-0.4376033038501368,-0.1884671898562846,0.02904793644809686,-0.0844328636004732,-0.3656827136285301,-0.2704121723773885,0.08646181213725503,-0.6612211069383419,0.6185402643990767,0.08213354347061089,0.2414491704746589,1.233731177615704,-0.03001699641602993,-0.8409071939124576,-0.2693592189563213,-0.04490153043742513,-0.1635703848165657,-0.6306127723694245,-0.1930812811402321,-0.1026244464422935,0.2730497555856248,0.1232975165795743,-0.1954199512380579,-0.6352171003789678,0.04663637220031239,-0.5913517376015731,-0.1158564380225022,-0.3137534769669005,-0.2452736981672356,0.1415978850294843,-0.01731337020186284,0.1433290377454928,0.5407326651598831,-0.01471203017459794,-0.1328491260219092,2.751104187654946,-0.4167619775289305,0.3736187848775872,0.3586010073358053,-0.4053478516551109,0.2166498084606649,-0.1061950889823554,0.4871918962732795,0.2156266165791853,-0.3131160866938347,-0.3313964784472896,0.04839947853822228,-0.625649337597411,-0.2940143778356702,-0.2920582285128175,0.2261617872228431,-0.1911673863687749,0.2347037792883125,-0.8543374708292504,0.1176024910214248,0.1706540917589733,0.3666992274328244,0.1013900689923063,-0.8082275420261398,-0.8412330133894007,1.125430809131376,0.6334458320031283,-0.01581543495016364,-0.08738831718797874,-0.4665883286267534,-0.2835910757143056,-0.6229574873390183,0.03281456139812863,-0.3390054173576527,-0.1223435862321881,0.7374093392755638,-0.2783486104234507,0.05309769427745683,0.1334382754599411,-0.3609989651526663,0.2407021411880232,-0.2293992659691196,-0.05707927961970289,-0.1522444595417921,-0.4479644873850132,0.2321616705163744,-0.3273474880346777,0.09788521667672849,-0.09244163439624566,1.267164507255875,0.9453603303408039,-0.3734040845342657,-1.738668061093355,-0.6312754002001451,-0.1864029830466425,-0.2082389582570809,-0.4058892074249044,-0.01990241155900359,-0.5042455729798249,0.6012412088196503,0.3264526235023588,0.1381195721670047,-0.9413626956444068,-1.128895579101253,-0.5714073279509142,-0.4443437827123886,-0.749022721094903,-0.7356942116242463,-0.4593047301454677,0.1564851499319472,-1.411680351628638,-0.5513226964702701,-0.3337509432559104,-0.4720422359580388,-0.4771328530218615,0.1739477647547963,-0.1991837197068945,0.08483524444703849,-0.4031638136541276,0.04000883193877654,-0.4886241647387612,0.1697268697277039,-0.560903320091142,0.2246398316880849,-0.06661680695905868,-1.32253746283356,-0.3189897546384683,-0.1599493054741626,1.887270579254515,-0.3990596013710493,-0.2330438336304637,0.7942325493733977,0.4308460315823192,-0.8819826117763233,-0.2601420207796449,-0.8562333158040938,0.1783370608829612,0.1484987071199186,-0.04126791567305762,1.450456802920994,-0.7733082735899109,-0.270044373467218,-0.6543371163020393,-0.1734448083926564,0.09773939807669353,0.4268865162546891,0.5865684462471714,-0.6586899279677725,-0.4202694193689601,-0.6603522056511689,1.022146867627803,-0.1749290169147802,-0.646842317340117,1.219380626318857,-1.04124870093175,1.555495841175267,-0.06027288965770502,-0.7663630045049291,0.05478319083919376,0.1223383504961683,-0.09298834529299098,0.08249149666889258,-0.1904622669821725,-0.9180362045454358,0.0231225890792718,-0.5734235498439529,-0.8826402407226371,0.02146987944797076,0.1512115452455139,0.2087940653227293,-0.2552011673501184,0.3622726528700681,0.3236515291166391,-0.1521219840131699,0.1777233526740155,0.3142828556282077,0.1013520077983703,-0.1937529206271163,-0.1039948029406522,-0.03476161501440642,0.1851664529910486,-0.913041715864547,-0.3785026039287396,-0.189840000426562,-0.2216340908311358,-0.3280231074115705,-0.3132572470287603,-0.02622157577213669,-0.07636489363905544,-0.7957377911591422,-0.1201662332447416,-0.130532835590253,0.1753382807489928,0.2657556303603202,0.8126542615753584,-0.4204966342777857,0.3562165686748771,0.01159541421043794,0.08246252762357927,0.1285238143060397,0.07834863650508135,-0.08833420397332439,0.3530725059451734,1.526352587995444,0.5141413607848597,-0.8510158760856243,0.3426209850696169,-0.9592706534506618,-0.5026739766019811,-0.1386502339075203,-0.2495345878927202,0.1734196303046468,0.1664263211850537,-0.4046223761321883,-0.3426432349076181,0.03336403968203625,-0.2134123775384435,-0.3158934714207106,-0.5812333043565286,-0.6186933796506048,-0.669371249344758,-0.8793955498678417,0.2573087619219976,0.1454938937191436,-0.9296652249504211,0.4228653695084904,-0.1500872161468388,-0.09948785652885216,0.1551496152392637,-0.154921874853303,0.308763848036448,-0.3898760598830138,0.2920276230938895,-0.2321805761693966,-0.7483021097197656,-0.4367940594203458,0.6681630991283185,-0.156925926376089,-0.2150151109591463,-0.3874505020941854,-0.2446831126132512,-0.03111753009138208,-0.2041639249518947,0.03533937980901433,-0.1123594292014586,0.2011482493118956,0.2997544148935407,-0.470592672164805,0.1399916057636065,-0.3204802394546406,-1.018894186351578,0.2599299747247304,-0.1501882158137483,0.4268070849431445,-0.5305235696628519,-0.8468227749885575,0.1777339319151387,-0.2578792862479711,-0.5102699636876904,-1.070488710257817,-0.3725376825921284,0.3582091254707228,-0.3563836310430687,0.07063889793363412,-0.1710352843958217,-0.203275741419879,0.338595588174479,-1.027400891997733,-0.05908509941769809,0.1894818136607914,-0.3005154880740775,-0.2338597313376715,0.2612352940767855,-0.03535879793342757,0.2630193088162235,-0.08476852977290732,-0.5081108757734722,-0.4989394449506139,-0.5529278522945094,-0.1424580730384803,-0.07536306449375441,0.5922445219630436,-0.2085445102809029,-0.02672742788210991,-0.4681598814287224,-0.6130000005345156,-0.9171750839516121,-1.206180167962279,0.05299499833847401,0.6145841083129914,-0.5144253248697996,0.08435637203453959,2.606476098419476,-1.609773245688368,-0.7192462835084261,-0.2761361958863552,0.1756017800394066,-1.399315640846306,-0.2993014906809919,0.2528825094416145,0.03497084327121493,-0.858441809294899,0.2044492338888554,-0.5215817352113455,-0.2802790285585954,0.1476830692929412,0.04913389700791553,-0.03993649140536713,0.06852428854748009,0.3670356285047923,-0.2177717685737603,0.1316681006700273,-0.5897337171895199,-0.2228701084432957,0.08397984981424264,0.5218573995942651,0.3219518350544602,-0.9295444996319393,-0.3473544015545525,-0.1131229336981676,0.4228077321158777,1.494424871447695,-0.1833868396560312,-0.1579657521900099,-0.3472482441889875,-0.0770506635066977,-0.3127692488642752,-0.04334492549072765,-1.063263120754186,-0.3679475767132443,0.1324159956986815,-0.419309995299526,-0.7722047641022117,1.745693138106567,2.058313960569078,-0.1848362768723357,-0.1567550705629335,-0.02922970777347937,-0.2645101828068352,-0.538438681185527,-1.026697427454793,0.06188311617823144,-1.17228472286939,-0.3500490234599049,0.1687681118357319,-0.6697257884481258,-0.7463612890909638,-0.1639260972034501,-0.2006550540710241,-0.2363043934083899,-0.6045995959610783,0.775842517043752,-0.235900270906845,-0.7453804806642931,-0.4030291087295672,-0.5747415221168407,-0.5470441646743656,-0.09659876573994453,0.1548982189134804,-0.5690768237131039,0.6211377683434827,-0.03175509276236467,-0.2155831505078901,-0.4744316148922424,-0.4064857407678034,0.2787640200416644,-0.347005648812036,0.8826581211906891,0.09277003254873936,-0.1333232778175194,-0.2862271322355215,0.1124471918636866,-0.5666860486472598,0.1423143407245579,-0.23617739525651,-0.2588132018266604,-0.175827892588896,-0.2123232114943911,0.03583821386389513,0.05242744212901043,0.402726784532922,-0.02687218355407673,1.055373167944064,0.01656220130582125,0.1415753035375437,0.2852877901100512,-0.4239416681094652,-0.4475369355504252,0.238371611251477,-0.4266589395818428,0.3576368225876396,1.649718031559831,0.08348716904369978,-0.04282334373645623,0.1244952458300497,0.3882716235785448,0.558841645142234,0.7179499933630527,-0.582983630943825,-0.7312487741269676,-0.7485952781884383,0.4367022932018779,0.001177054083340005,-0.06553600841700798,-0.5854022744554964,0.01961634817854774,0.008590657047295988,-0.576383366102186,0.04191816707726126,0.6020595568540021,-1.37657441302184,0.8749910856028462,-0.1531220932602076,0.1577397527414672,-0.64831111346972,-0.1627102442118232,0.3328837832993461,-0.3893059825742709,0.1035377637212709,-0.2798036963707495,-0.5010402316837336,0.07144138257381646,-0.2929667974177725,-0.4303582556468891,-0.2704827854883256,0.4238834749065161,0.1409037736608616,-0.08902982478026066,0.1546208308387359,-0.218358104441288,0.1112433581656696,-0.2043388795453662,-0.108869747004798,4.299808941780292,0.6592817250157538,3.05323344681716,-0.01742079795209182,0.6382779508007262,1.172867304345544,-0.5094606778154691,0.01260753373448062,0.2613730599406895,-0.2614700942153052,0.206309382694595,-0.008340840982373074,-0.5241334400295966,-0.3787284974330244,-0.3923323183329413,-0.3354488877104634,-0.3696927419750884,-0.06117110731054003,-0.2670599250936528,2.509721644304148,-0.7029266617026357,0.2591347381597633,1.410238522791178,-0.1706848201171513,0.08936931392311759,-0.6184950981418005,-0.6848393747071773,-0.439910587887152,0.8336720568051431,1.172630541120824,-1.185635522050692,-0.5980821977581992,0.6693909371097285,1.616564956244307,0.1045956599849833,-0.9417314482217397,0.4425873238718953,-0.1259278476511194,-0.3895376864373063,0.1617694417096638,0.2742955592619031,0.4698257009798316,0.419705268564234,-0.08237187422036388,0.7795206236634219,0.1564283018980041,0.0512487672681056,0.466954647531525,0.04953294232803429,0.1830464417572273,-0.6882319211445392,-0.430347258461842,-0.3121494123338496,-0.3020011929649196,0.2836107035338068,0.5818364476436589,0.7744345102679426,0.03052091791802311,-0.3907198490868635,-0.5417443314907144,-0.6775779918752897,-1.798255455494522,-0.6045515850386336,-0.2847308727121127,1.543266485524679,0.5027470240155464,0.6203979459566122,0.02451440521367582,-0.1697711458107425,-0.4994934925700445,-0.3404412438811732,-0.06288600662389698,0.1484036642025365,-0.01192844423948127,-0.4158662136123276,-0.2961058832586569,0.9543370407887161,-0.07752964840556367,-1.12855172905515,0.2520246045023679,-0.3698826311577131,-0.3210651266944441,-0.227248577561814,-0.2331853032145798,-0.6013998640090394,-0.002873805760195565,-0.6174742437705379,0.1156118074176519,-0.3073885156314102,0.989231320787151,0.07412112469398202,0.2911163234078994,-0.2570422065119633,-0.3586496591303869,0.03315811804350009,-0.02621133150214171,0.145028116507044,-0.08134877149672941,-0.03666076333586048,-0.5340559282195326,0.1948105992249562,-0.3319391332881388,1.818219594793237,-0.04832219072416935,0.2617634950151111,-0.1347084031114717,-1.273533127031598,0.2780855759448168,-0.2025534917759527,1.157420732624145,2.028028218955292,0.6636911655801933,0.5733169706091603,0.00216378657145755,1.558947589264757,-0.09510186229832418,-0.247668163863053,3.311512953731639,-0.7000004714286284,-0.5068748545534464,1.061977398033557,-0.03942652114139177,0.1711955546450388,1.127622640545972,-0.06771726636775371,-0.4588471927698429,-0.9268509598133232,0.3388934813525273,0.1145057264748184,0.09590931827250594,0.1883818725016548,-0.1568531937714025,-0.3361936340776895,0.1624909322914364,0.1623440552422389,-0.1000908162369205,-0.1957813149590993,-0.4444445457088629,0.09772700401233944,-0.5535720194674424,-0.1851388667994868,-0.4926185279439105,0.2156609758364181,-0.8555063327728305,0.5406000492076832,-0.1765583222658453,-1.064308558298127,-0.2741948806864462,0.02024432529336999,0.670306040120868,0.2531186191970818,0.05187838377967989,-0.5128416569491505,-1.011166762902108,-0.2861752272213005,-0.09498184780952799,-0.3509263752741199,0.107644473353986,0.3781421930013572,-0.9926668341989542,-0.4432846341398515,0.03054649744901685,-1.441226846193652,1.01532598312743,0.280084076576391,-0.8428996204690486,-0.05675154028094794,0.3221206896411983,-0.1128148064375923,0.3339839388051062,0.1177984102144259,-0.2945960050313162,-0.3508127984910598,-0.2165677679798887,-0.6149362520187747,-0.4384491093009537,-0.010138654694441,0.1496011975318133,0.0761238832259355,-0.1391202442887555,-0.1023814167645169,-0.3782784070918611,0.05752715991960239,-0.8056240244482371,-0.1070549004034555,-0.6493315925880365,-0.3833326263527009,-0.5654447013915868,-0.600862126545794,-0.3939283217217584,-1.257306565823956,-1.310295458523461,-0.770284933761939,0.6284467451314046,-1.174302146120531,1.400664929797511,0.1295573128826004,-0.883869477713663,-0.309323606706734,-0.4217507762120989,-0.4155036417408404,-0.3517523081295268,0.5657885215261602,-0.04261050831034449,0.1725350187747932,0.6861952786867386,0.269637926149935,0.2192882425774242,-0.1801531654169165,-0.2310825702966605,-0.1765658002674535,-0.1795470900081074,1.542429165111641,0.3984209833295941,-0.4119239661835316,-0.3074305107284033,0.07548811489335706,-0.4185602276318051,-0.6846829319868446,-0.2546880555818914,-0.1193705975379043,-0.5422298295443102,0.01757816447547093,-0.02108815896314055,-1.131818804854962,-0.3954578237614444,-0.4350494790034305,-0.2568089427197582,-0.006264597018048199,0.4473969287926253,0.04263443523973603,0.3571991475133888,-0.3218047195169944,0.3651819365037394,-0.1006070663452832,0.4834251967016735,0.345186019411421,-0.3752521300439752,-0.2287518591721016,-0.2614012629042324,1.556945346797219,0.1850182418411978,-0.2865634076457053,0.7878746590656548,-0.05226456101953217,0.064193598267081,0.1299218788316908,-0.1630280353788375,0.2267959045407229,-0.098140585932442,-0.3572521036042795,0.04519582044929653,0.1926753035780405,-0.2647556734748682,-0.1753602181824358,-0.734092945581959,-0.01140766671679392,-0.4106223827811941,-0.2654809750112865,-0.9595719536007757,-0.4915821077750682,0.1442911908603851,-0.405301373576303,-0.03020738724534266,-0.3565149260894228,-0.2272938983555074,0.1258027856483041,-0.3170930168377668,0.05869252954594713,-0.8091702887796416,-0.419619949641847,0.0920849803303684,-1.038983705596935,-0.6732308127151441,0.05937580997606396,0.3683357688445553,0.4831506016481411,-0.1846454301815181,-0.3849620917990633,0.7181749576793637,0.5275527069936997,0.2340941231568394,-0.2647647238495869,-0.1230596607624597,-0.5037013911921343,-0.2822012712852667,0.2193313031419828,0.1841759098260895,-0.06570334834307905,0.08943226900472018,0.1767073097571022,0.4619526359407943,-0.2866907389864307,-0.6259593095746201,0.01031010410767223,1.007568657536195,0.01403406834160431,-0.3417129284056133,0.6790607445839744,0.08435113494923489,-0.4101518239045157,-0.2779602063157967,1.461991278728913,0.05456301794373299,0.7797603606774198,0.3870131407126414,-0.4445688748878723,-0.3895676143295426,2.055546358698137,-0.1502128020410956,0.4155942612399297,0.6286055657691241,0.5900404872044246,-0.01561760717059856,0.1004835549790694,-0.2502389752444027,-0.4578326970323688,1.919023074440866,0.07502141658722038,0.9345748583528068,-0.4057292673984484,-0.2346421698668845,0.1418935585947491,-0.1705023725609243,0.1652679837803472,-0.06956466220713733,0.1842974639458481,0.3748337304076022,1.685354104204078,0.5761346040633696,-0.3908870811074941,-0.6124493194785201,-0.5130311461195962,0.3890048738063253,-0.3523221081974913,0.2962633508193386,0.1301862809604366,-0.05224597687408658,0.2375835788013299,-1.316849525762269,0.2783373322769044,-0.3254039025847433,0.02691108664799358,-0.5907146240927573,0.1492010779102126,-0.4639492111092763,-0.7476585616776642,-0.3459511029040037,-0.303374779232902,0.01986070607754101,0.827570689625912,0.2659873130356646,0.1710889818510574,1.55095533734798,-0.02209888320440813,-0.651529227455554,-0.3642397683837459,-0.06536253696869705,0.5063691531484243,0.7046449127589071,-0.0255039894608672,-0.5309371786110737,-0.3888592628171745,0.03045134881116208,0.9162814972007858,0.09435204260235326,0.3467780282802193,-0.2671742313367388,0.7966412879859057,-0.1339755897318569,1.131737494766003,-0.7367183370177163,0.1650813682933473,-0.1576499212120114,2.245869142281044,1.692723372130982,1.098180029883975,-0.2453881688874509,-0.1384465031230218,-0.2108024501584801,0.1125119547283891,0.4127526364996712,-0.04663907390035384,0.00641257088517738,-0.1022025506027396,1.218050295684189,-0.3399712521951385,-0.6259965484501626,0.178873549685206,0.1090020811930178,0.03194408450334547,-0.3556698449704296,0.02269351392242899,-0.0606527084179276,-0.1643066049792947,-0.4771853352547633,0.01679601264989113,1.395232674066679,0.09169448525658341,-0.2418249943079384,0.3382286079688839,0.534224255121036,-0.6756938519805522,-0.2774671896722215,0.01393268468462697,0.01561116974786747,-0.6198923440200295,-0.1770034767235708,-0.7994643108519952,0.1646769268897599,0.07429847191549611,1.337554675236588,-0.3486595762391138,1.027241419035206,-0.1266430517265391,-0.2368093371816963,0.262058593662497,-0.1680428795885573,0.1012912433029975,0.2245426593893026,-0.2174775834717977,0.03376384079301454,-0.03402559528988665,-0.8700060597104415,-0.4282105055515097,-0.2002968545682023,-1.14876935771216,-0.2635566490052338,0.7472127473491292,-0.3195494117305848,-0.2255248207729768,-0.1966999161166165,-0.2698979678712698,-0.4447679320062266,-0.6541733977634593,-0.6376754244865701,-0.08123004501852213,0.2663525102113315,-0.5245388638062005,0.05278788759260707,-0.02385772234854439,-0.008229506258641791,1.824633906590876,0.01779626013513724,0.5310105603744467,0.3918031470485773,-0.5522704821191687,-0.8217931016538605,-0.05058482659881041,-0.2460445539309503,0.07809446960530902,-0.3452084700074935,-0.07412559184430849,-0.2356074879579748,0.1937897863820957,0.6428269486517291,0.9895490183347939,0.4570353829524453,-0.4081089694160276,-0.9416726698695271,-0.5124593708980046,0.8240244289868641,-0.5884816157635648,-0.2604612865152684,-0.1910931063839769,-0.8164435290058318,-0.02233783322677872,0.01771867087197016,-0.1816711063214159,-0.5523503897181712,-0.02320786552249056,-0.446056393989114,-0.2270778686268171,-0.5449931026332397,-0.6114554583849069,0.3828473291045283,1.029398747716894,0.3491143977556905,0.9592084078119613,-0.2336460025413419,-0.003239428296384968,-0.6988501742972921,0.02102288880171825,-0.05049102165054675,-0.1273465335857215,-0.5586651857783461,1.759012542884044,-0.5962311056502407,-0.6625682165095764,-0.4569261935957652,1.036033727265829,-0.5502332643327589,0.4823027302640613,-0.2872325597808156,0.2935251283552393,0.6431924349940101,0.7197535670770986,0.8965567521950776,0.3105208196642114,-0.5187591531494899,-0.1974652060569129,0.09875590439643589,0.6975314168509836,0.4591469272230663,-0.1686470782569316,-0.4847407194080205,0.08797529470734859,0.2640344106296585,-1.471067367504321,-0.1876521849403236,-0.5253091123701539,0.1731767220404293,-0.1610677956831423,0.9666843688073343,-0.918901788192965,0.4025733374537482,0.5642707039498767,0.2072719708527108,0.7386143609011003,-0.03491579293194725,-0.7692219034986311,1.152665777812416,-0.337575602042277,0.1092070557093515,-0.3977743039037977,0.3356470264826056,-0.2964377137311031,-0.001878934858159372,-0.5114286989642647,-0.08304467937633819,-0.1171412143898727,-0.566205314528727,-0.6737355067530452,-0.08884079454596285,-0.2928233512561688,-0.2161387805958045,0.5691001432828939,-0.5516434073680567,2.216540463664688,0.2139507959670553,-0.1754074577055381,0.0242426658940902,-0.595331629881502,0.180287328751299,-1.155079566954584,-0.5058262982696338,-0.5395928084921542,-0.5954502184646181,-0.5719878932681367,-1.287147034822638,0.1688437045323186,-0.2953497668327159,0.08673287665106144,-0.1199426042690648,-0.1452867482523927,0.857948800559833,0.3569443330471923,1.043519618768939,0.0764370089345193,1.505166883528455,1.934941051177649,-0.7870885371063,-0.4047567694586387,-0.004013728833504765,0.8445578857077468,-0.02049169829650433,0.008429417784985111,0.4275292299901685,0.2334504563024911,1.375028780507735,-0.5056159188984359,0.7414233799831736,-0.3917695674252006,0.07036979283177953,-0.2535602458357729,0.1391046208720365,-0.1972735302016835,-0.2372370428701902,-0.8038522747778586,-0.9766254739997003,0.1793363557970092,-0.04146320912024393,0.3252044663766226,-0.8364135637288697,-0.3928738672141563,0.2381075512527913,0.2795857412295475,-0.1509755056887427,0.02449092759670777,-0.4729227312676114,-0.3150789640641403,-0.475109997193193,-0.1937816014141065,-0.4037272813411361,-0.4304628127841671,0.05614728263005556,0.3351495963545049,0.08450526615868902,-0.870843727166555,-0.4073691103360106,-0.6578876605477268,-0.4290577528353733,0.06624532656735317,0.808597516846933,-0.2533727988035948,3.16785124765831,-1.338957956994168,-0.1928872753452165,2.5245819508104,-1.053650700347246,-1.236511821253072,-0.3153254977043157,-0.1248706826245001,-0.8710522646865885,0.1130834558998972,-0.07736239526626015,1.451751017797896,0.02480942184075916,0.9578829252510476,-0.8740304664023459,-0.7175321432740143,-0.2247057290242169,0.5378450450184532,0.2615225903528746,0.1035140435085736,-0.2240572772835246,-0.5336076001301616,-0.367588259511081,-0.06634122326415581,0.0791024682758104,-0.4114876260876364,-0.6966683704811957,0.2684109152111926,-0.2666607372805248,0.1550448879642452,0.06756976288988401,-0.1748662936921419,0.06574569005707909,-0.994439459949928,0.02437629149339206,-0.0924774125980618,0.6532360556504844,-0.6171692254105355,-0.3771657340452175,-1.261557519589612,-0.2120628648504586,-0.6114410052246341,0.7718680520914076,0.6960351185526332,-0.9808939041836872,0.5251813912989728,0.1509610501780083,0.1543592712202418,0.5159594035793896,-0.4104916045866877,-0.03205074200565604,0.3666776159787871,-0.1365684711319344,-0.2995176872154067,-0.4448464640515728,-0.4414262610920369,0.1018066917059996,0.4643576716588982,-0.1572861496157364,-0.5899213556553456,-0.1289070256946628,0.00399301620366993,0.04418618683861599,-0.7068346363942253,-0.3755704819438235,0.2175264265998667,0.2038099961027201,1.868861833619835,-0.4466959679270662,-0.2194277271820247,-0.3522602334317774,0.03173698999878026,0.07663576410840356,-0.2746584436216894,-0.3931315639387514,-0.9270161948550595,-0.2873564180545258,-0.4208392381800745,-0.3226624387966471,0.3418630632329242,-0.3033076507720946,-0.1378099782472181,-0.4886754940630545,0.1185338048040969,-0.4356261090610949,0.2558010507531209,-0.07875021307280153,0.1188881456047625,-0.02238445290851516,0.02335920579708877,-0.1509604582452977,1.600598300277001,-0.6045649902512846,-0.204782400009522,-0.6798302430106674,0.1751267421988598,0.4700534394693964,-0.3554013560715121,-0.3789852102476731,0.392559877997975,-0.3455770828374695,0.07133325391258528,0.6357085707742371,-0.8798934185987365,0.7544769747118836,-0.1109959904410164,-0.6372354287370801,-0.4617949107083254,0.1094332098420092,1.869335412246109,3.488480907604444,0.08092711110829372,0.8862335444947325,-0.3745943922239876,0.007505844290076563,-0.6039094994743025,0.974912980719051,-0.2745785255328121,-0.3397918181273094,-0.5839122786486501,-0.1695709930626771,-0.9733385965388288,0.3157014517442452,-0.6607164843283131,0.1794497602030559,-0.625469799734107,-0.155376792996606,0.5178153207925295,0.6806722957412982,-0.3652626245703255,0.009781724721631326,3.520790688037854,0.2727480115635781,-0.07903914702811358,-0.0980313967545634,-0.8016901138520768,0.1205151723911904,-0.1527744309198814,0.3527691332017467,-0.1756492575516307,-0.6191635843009293,-0.5949082521762106,0.5456923118104819,-0.6209122728991769,0.9495827162937979,-0.1877836922332688,-0.3916838301047645,0.04349748202713671,-0.5529070140212974,-0.6348925792938623,-0.7576330250096613,-0.1393928457519463,-0.1628006734601856,-0.06781359319336719,-0.302491796602302,1.556727483075941,-0.5667780177941043,0.3781205266395473,-0.0567711748617373,-0.2147485243408726,0.3369293099775857,0.1434066736335177,0.7374469368852455,-0.1157681931764419,-0.1346104247609426,0.3425446316381472,0.1206232168298364,-0.9027418770299347,-0.1878955495991392,-0.07817057089454124,0.1052255104385131,-0.4679465847338381,-0.029419183073257,-0.5007376553360393,-0.2804229927798013,-0.2869142753094931,-0.2216251814059602,-0.4733683356227879,-0.6550435045229762,-0.01930235248706122,-0.1032775333707567,0.1790052290624458,0.06515277484872678,0.05496616380209399,0.2299828762693651,0.3571977764936403,0.8301071236982775,-0.1143776809418367,-0.3494674586228743,-0.6313109972353445,-0.3486063828501358,-0.02558705943271239,-0.02450454287460546,-0.7191667138002747,-0.04156857223052351,0.7902125903843366,0.4485369762394234,-0.1708632266232072,0.9381684278473129,-0.4061536383181765,-1.215521140069289,1.143449653373236,-0.01143178049545704,-0.4705556632582399,-0.08800893206251914,0.1856867050198282,-0.2834021348263152,-0.1480802654186188,1.11945475986267,-0.432382112863176,0.5805053627245601,-0.2756348519800023,-0.5963144798188486,0.5692448529966637,-0.5134019726208087,-0.1111084012431381,0.2474751462887123,0.3877104242181399,0.4380023718729795,-0.2059910054583092,0.0839598241784132,0.08878040972313715,-0.4496410665600707,0.04784832108758457,-0.2492751282448496,-0.3407212956361003,-0.7993289018154122,-0.1533231687129981,-0.1983187822285891,0.8813274851234311,-0.3363003013243054,1.888473141430288,-0.01226741231886166,0.3098581185818898,0.2176035689665405,1.276911675695448,0.1653523866275152,0.07377732093192683,-0.1976886465902066,1.03028779974026,0.2944083107674834,-0.29679920237535,-0.1867628376798777,-0.05131823817828482,-0.1299310566983645,-0.2972883600961279,-0.5006201946136575,-0.3906793911417236,-0.6022865732568372,-0.442395668778159,0.4111617169685309,-0.2317253447087725,0.4342402084895505,-0.14265730774279,-0.7343632189785808,0.03097500831864932,0.0987842375615762,0.09349625491414093,0.4043864176811259,-0.1322840773398148,0.5144742810599967,0.2899490955074739,-0.2107624947538915,0.2027330598715136,-0.4540926238293652,-0.5732155348017192,-0.0008478801676210527,0.9829924179898322,-0.1518896793645072,-0.1893600442768571,-0.7218261558075175,0.04154944342260494,0.2117918173900431,0.3839038211791662,-0.2341612986377064,-0.2617978828685287,-0.03271400405325182,-0.2504757256696261,-0.2396386045446003,0.4543119389856931,-0.8070003051105726,0.1822620235316509,-0.3891837902701777,-0.1802858808054707,0.1735729906288805,0.1014960505496951,-0.1870708109359009,-0.3055806120153391,-0.04287046379862692,0.03859176115744919,-0.4757943117249646,-0.8696345540931232,0.6807437302151912,-0.6502106106814204,-0.3205333143465156,-0.9788895553067185,-0.9481823718434097,0.7575437135505351,-0.6869046897035104,-0.2934102419877494,-0.5593697098957556,2.364895413037688,2.894197481317691,-0.1662335764449367,-0.03843034226492922,0.2094523708412655,-0.4162030475163533,-0.1530512593419873,-0.1148295479488962,-0.08546709675161118,-0.0930076989324415,-0.09239236632569661,1.695407703852649,1.11438203865111,0.6780138342360045,-0.9323317564550918,-1.007292859929083,-0.1740842038222257,0.7026489069389782,0.1216929832308189,-0.2532270571075073,-0.1517635931146021,-0.06336373692796257,0.08724616308159237,-0.4011175996594293,0.1215320624608461,0.04653234903683136,-0.1053027618571215,-0.2768916604454812,0.7348333852209922,-0.1398484204292365,0.9182453565855591,-0.3925282359663579,-0.05915087482023332,-0.4283995658355468,-0.3646112476465757,0.2067906967408779,-0.6771233459286035,-0.1919099335037052,-0.7166003078200162,0.6458537326095161,-0.511132220329033,-0.525255231703305,-0.1225694534833731,0.3303999301198901,-0.0388378268615085,-1.363935966795747,-0.7057380941081793,1.816067021887769,0.2168868874915217,0.5300809044472993,-0.07959945802475857,-0.5849244241302868,-1.3181796940687,-0.4762047900302691,0.06523973738653989,0.3881089636111369,0.8728629773639164,2.359805195906269,1.094310097907425,0.3226336415365276,-0.1282237485762351,0.2041076523217256,-0.3806399750355224,-0.9234886418173522,-0.06700917411984048,0.7671790319409922,-0.7137924372022878,-0.2755747976170424,0.07850382911437942,-0.09480590984631296,0.4733611150647744,0.3765766550872012,0.04935920847689713,0.04655232629238124,-0.1573808477624625,-0.648880429737302,0.0372574889851643,-0.2774493701449075,0.1613003348093728,-0.7159940638916819,0.3741955281108402,-0.6975889610104433,-0.2253345287946643,-0.1523391046636125,-0.09181753603609254,-0.4845650069846044,0.07810663149946381,0.5453780531934411,1.156303583099432,0.3520615945878042,-0.1140338307110167,0.1440762579234396,0.06072340137276135,0.1573583638566292,-0.1814080637771068,-0.04451725002648221,-0.4039051789342084,0.3127892737051442,-0.2749936200644656,1.40597155234813,0.03107044101045588,0.03091733709174,-0.8549717323136961,-0.4945413065390755,0.6569365692941498,-0.2072959878824627,-0.5494477677116025,-0.3573307046932578,0.3466106983287185,-0.5584949688520499,-0.1299176095224449,-0.0221110747685854,-0.1477876463225567,0.01842764792170858,-0.003943371748936814,1.775423350769416,0.01805496336679854,0.118069286964826,-0.07357160648240268,-0.1755257283603429,-0.1880912085242582,0.05075821292111495,0.1139260282166441,-0.181132662554884,-0.0668487894079221,-0.4106048647491807,-0.6273829554032861,-0.0414850011728987,-0.1179853824967093,-0.1362552924741937,-0.2467422088272421,-0.4235065595171976,-0.02309122366733147,-0.1578850927668756,-0.2784723055807173,1.042112876644733,-0.4570304415377882,-0.1702275606971752,-0.4753831624197793,0.143657218038408,0.2513046676298323,0.01314010750443598,-0.03786625030199333,-0.4887227092518895,-0.3318053417858832,-0.02402317140210293,-0.3603485633933907,-0.01815672503030081,0.5869113443831264,-0.8773126424687849,-0.3204769973124035,0.2145790555996682,-0.04732561612032829,-0.2901596806585403,-0.3445599975997076,-0.06013744030555327,-0.2394793893858731,0.2294218827807972,-0.3221418553335584,0.0612072887998887,-0.4389345189842356,-0.3293170691202741,-0.3668872332755977,-0.7092009270589819,-0.7182079531572819,-0.525206299864476,-0.1714491119068143,-0.1681303604090684,-0.5477863957741254,0.07704035931367266,-0.3604690590231187,0.05777483906317329,-0.1005406549765873,-0.02348826129669269,-0.1346919749888761,0.3127958448901778,-0.3111345157951768,0.3015486737354436,-0.1675684898080678,0.635325697830417,-0.08250423628674215,0.3023978631413662,-0.5108097761711796,-0.1621444762459047,-0.2615315274624891,1.1147879666145,0.674777590201103,0.2325299169614269,-0.8550153448744634,0.5196675506260882,0.5410012213597921,0.3627501543553977,0.1217669452697424,0.8096176844306913,-1.278368581406409,0.399337129191544,0.09346642480733643,-0.1827142695950463,-0.0171999812551899,-0.2897057351807409,0.154009796928275,0.1121918960331738,0.1821481401906699,0.4310050039194736,-0.0932304134102158,1.286383578673224,-0.3744011660110514,-0.5841256681700562,-0.29825832867193,-0.4406390943068136,0.5368658865203353,-0.2359764759057706,-0.1604317625630302,-0.6880590431110196,-0.06064703535173368,-0.3179731289799204,-0.06585793433419158,1.334204656560751,-0.4836661567725761,-0.9749427518160118,0.6386530049474354,-0.1422355545972368,-0.60228429850042,-0.1923202580075347,-0.3850966472476633,-0.02055933828489997,0.01437636400309441,-0.02907620742322899,0.01487228997065867,-0.4831416057261631,-0.2472513786689222,-0.1384526289183989,-0.6414610607541502,-0.2509471252761236,0.1190136273318299,-0.1822507168630399,-0.2418057239747266,-0.008241709825875486,-0.2174052685694099,-0.4377274422689765,0.1901520071848624,0.05685467595537894,0.6909249918365706,0.5940444325639026,-0.01023722864163414,-0.08084173228287768,0.1444678967394543,0.1826326516562653,-0.4645152300747961,-0.8623654061591122,-0.4765716345650409,2.144551694678404,0.699286879749571,-0.9984160240474147,-1.691039549973426,0.01363494221539056,-0.6770315471710853,-0.181364948723657,-0.2978975968564722,1.16327069876918,-0.009902151557135778,-0.3721947835106859,-0.5374830968293627,-0.04009736062340823,-0.36860096232786,-0.09485499795236418,0.6231805306087613,1.16202107147193,0.1044328844501528,-0.9395532581006545,0.3126941275749798,-0.4053747032559576,0.3177592549760997,-0.1230893628634531,0.4137213522951809,0.02073404982109909,-0.1413696255723166,0.5426210351928366,0.1820973900145856,0.2543893143753039,-0.1585513844080573,0.03884886246108114,-0.3921509242571969,0.06921053500815619,-0.3639190644109743,-0.07804137301919099,0.00928933690977885,0.7055940471475491,-0.7956973905498295,-0.8215562860065602,-0.3805864002033574,-0.4136261993640764,0.4942665264890224,-0.1784426818961354,0.2013080821661755,0.8229057265349931,0.3101570731972514,0.1941625672633745,0.6212582540010644,-0.5931358021332682,-0.424008967885676,-0.2054732347220938,-0.004652594269321132,-0.7716863412367883,0.5621164137350037,-0.3049226955341699,-0.4038446127876523,0.3962412821473023,-0.3522639878826022,0.2299650054896269,-0.2071831181774676,0.6720558294966059,-0.8368768965546595,-0.3550141420696932,-0.5397016041359828,-0.1394484797563554,-0.332528083026265,-0.09162808363160296,0.07868657018138259,-0.4309078805279524,0.1579532768139208,-0.1259553101119474,-1.111589414214144,0.0349795307297729,-0.5031017323712267,-0.4372641792199638,-0.4305676036036935,-0.3611803783478654,-0.2551208746547304,0.06224538449074462,0.2426703478739806,0.3661176812424162,0.6100350137128787,-0.5985061354031342,0.04230458907510243,-0.5527903899214911,-0.6306254132822462,0.3685670851233976,-0.9259114041802172,-1.137049230272858,0.9486456959028394,-0.6963146959518223,-0.6788932470863804,-0.4418119479222838,0.01998790665007053,-0.5989395509794935,-1.321409069055637,0.09595907913043486,-0.08204429801560453,0.2729571576449259,-0.02464797316321354,0.6538207658887281,1.064159675338893,-0.5482105031647275,-0.08480135388600152,0.2548458799103857,-0.475284466070412,-0.05597786703528414,0.2572661579175906,-0.4736031989767757,-0.001139427102343688,-0.8032444633590164,0.5638129485029204,-0.4881387278730027,0.48189263391978,0.06279723455196862,-0.3472401280519004,-0.03886217620983073,0.5384508668138024,-0.399547338466608,-0.5527997578856539,-0.01226428431946497,-0.1146483056509591,-0.695639513389167,-0.9452126737942967,0.5754771002634521,-0.3358670570493343,-0.7011114068658525,-0.5646938077240159,-0.09436636906714986,0.6308831396965281,-0.7075476409210513,0.07653575688288478,-0.4619286270712584,-0.4038306568888184,-0.5432305518234138,-0.2426652630999923,0.1201380800536703,0.1877434411858363,-0.3985721738843111,0.4208594684041524,-0.4457567793920075,0.7260066065636152,0.07966533907375919,-0.1829965025144728,0.9028599528620528,-0.2179692073841805,-0.4377831997577596,-0.4666725032232089,0.2068489951259828,-0.01795225894864088,0.8537539363809127,0.1447921833950485,0.1023147464350304,-0.685613581971657,0.3406586789864752,-0.249059253243119,0.5518326404025456,0.4927470786767946,-0.4355545316028946,0.6841734504090253,-0.01895902235252273,0.02965991095866816,0.2008701019194122,0.1354810343343897,0.2124291056854472,-0.5208215499982634,-0.2813079513980229,-0.1045788162396316,-0.8803040178011218,-0.1793490081889243,-0.01657785276010574,-0.203491494991611,-0.3778894188124068,-0.5315527707962494,0.3775436167562337,-0.428983000120542,-0.1941263036040964,1.299220195210225,0.4715659217992677,1.093230960272088,-0.565497869041675,0.001054296997086023,0.2765098417699067,0.01933775644230206,0.1167350330481659,-0.8748755649734256,-1.622548323302571,0.08714441581826753,0.3990783965568611,0.3924489930914559,0.1727833472217856,1.111301380268431,-0.03984778603628695,-0.3661099807778747,-0.5351692015333769,0.7520622991911383,0.4673472790261906,0.4729630539713562,-0.01645982672130242,0.1890093586220777,2.154572767520937,0.0961066041166911,0.1704653138193122,-0.1189106641873611,-0.3488397022071699,-0.5333356731302813,-0.7059182946895575,0.1323921240174611,-0.0202061473928755,-0.07100911206795155,0.5116245199940538,0.02597138948017199,-0.2978558652700003,0.9002116527193885,-0.1357870126846784,0.01758074133915061,0.03071155328861151,-0.6509278947623592,0.182589709147498,-0.579158871490999,-0.02760713281225456,-0.5719793635189956,-0.3515418258852812,-0.7708261384435592,-0.1211326537219166,0.1082151016155605,0.3901135260654129,-0.2316363687343048,-0.01035301903424477,0.4870214368720405,0.1689765623250967,0.0449222749898557,-0.5709246263396993,-0.3678299023710517,0.3844030838968905,-0.1346060422091194,-0.2004584138943113,0.02930068515952482,-0.4331770456949969,-0.2614860971631065,-1.237600618537955,-1.22073661234925,-0.4739837711834307,-0.2991064977965529,-0.08489255509276328,-0.5409934831490479,-0.06816459573541379,0.6445389558234978,-0.4326069336372768,0.5003368610242495,-0.4694928749050359,0.7625773637153035,-0.01751264763180801,-0.1918035515917442,-0.575591651484889,0.0421996678233534,0.3418934863657684,-0.1316561273126241,0.07331498595034613,0.594776561605069,-0.4786983647626721,-0.2132669995778848,-0.3922087989136359,-0.224276768266631,-0.1874817011342073,0.0849242686048487,-0.3528565909178555,-0.2515112368341787,0.4362323040303073,-0.5836047909729474,-0.368827943902357,-0.3979136667706182,0.0001029694627884938,-0.2010115614238484,-0.1415154369856701,-0.04225053360404573,-0.1368281701996572,-0.4393158808976285,0.2063044092372789,-0.3835349329184357,-0.256988587063283,-0.245526877439797,-0.4033779318142054,-0.9829235870577481,0.08847661899217982,-0.3784885951323417,0.3228894335590037,-0.7520792752471129,0.9792833349798153,-1.138668512534032,0.1911751306546439,0.06102315370912286,-0.5041890620151305,-0.02709582215191847,-1.858032498494366,-0.863546603676886,-0.3668565113993548,-0.1213662776444139,0.1734194271305148,-0.06075711514835034,-0.1761975074433591,-1.275921088937393,-0.2621522717616495,-0.3303985820809904,-0.2207073401996186,-0.3152301308796303,-0.4018272538197577,-1.143753150075571,1.200172280878919,-0.5340677602104089,-0.7727786211068935,-0.2656886632589444,-0.5021090802616434,-0.05089231507538424,-0.1704095904089195,-0.003136775892638936,-0.1204879418249474,-0.2780572983432156,-0.3518048620366168,-0.6162683984355698,-1.151226666698237,-0.3010980562766253,-0.3598663149304595,-0.1844062204111634,-0.5360788967304316,0.1670314203376887,-0.2379135055728577,-0.5188643339940721,-0.2858885927980333,-0.3649682823910328,-0.1305621445341506,-0.4135933050842036,-0.5287547100903598,0.3378559473294203,-0.2597305296113599,-0.1824124691791544,-0.4593531457420161,-0.6739987171485907,-0.5797865932682534,-0.2235226902591819,0.4738276119521729,0.3516546688811738,-1.482157058366153,-0.0115537834193003,0.1180716996678134,-0.2965768297411798,-0.1907361520482325,-1.04357976715737,-0.1019754902708767,0.7171685384897489,-0.592441856349858,-0.08738865795376832,0.06779660462994402,0.05787715599966304,-0.6907793808977957,0.2642064448392913,-0.3735226730058134,-0.2570286524992483,-0.3928822367114598,-0.4864483625216404,-0.6401422817649886,-0.8561649801882124,0.4726805261821374,0.09816822726408828,-0.3158583368990913,-0.9752049108868696,0.6335424461085765,-0.1891761413635383,-0.4564671763522137,0.7828391134206484,0.6496169286701035,0.3263766067114709,-0.7587460913091667,0.1064130858828851,-0.3846371254868291,-0.9994320273117168,-0.4137671121089732,-0.03540890893466737,0.7568586416107972,-0.364786717693378,0.7062933402263206,-0.5294331451992479,-0.7989546999993221,-0.4082301624601676,-0.4741710833046269,0.07003121240717471,-0.191447844312116,-0.8492494075867757,0.2847933835597711,-0.6882506054407227,-0.009482214540612472,-1.000034084893556,0.1158611250418022,0.1242545299763041,1.013768266621057,-0.1254808761454138,-0.2630230660535647,-0.59055588733918,-0.01370262825743383,0.3076453425297783,-0.9442772408884907,0.431497290302004,-0.2426725088090465,0.3052731816350029,0.34902528322087,-1.136852951695718,-0.1377879475119516,0.2002225713495584,-0.2537607880775956,-0.6036786621763574,0.1525763340250814,-0.6912768258346098,-0.005349924524114726,0.01666369938975651,-0.7263359502982025,-0.6438925817719718,-0.1875388410601528,-0.599122420477592,-0.2486860696874831,-1.150250075752004,-0.2256945624084363,-1.119419621137808,-0.3301371242347551,-0.2734221448319137,-0.06926055541701889,0.06541652405003184,-0.7726413038630103,-0.4512380853019742,1.941390429001822,-0.13990458970374,-0.06687544068090974,0.7858824901643672,-0.04410795848331771,-0.215257797640131,-0.2926256438517795,-0.453798077960756,1.249182555158446,-0.8501638924181431,0.5044236623233396,-0.9728156805738092,-0.3471018146068313,-0.5001121311659482,-0.3989634159555392,-0.9864423619777922,-0.1458482759033406,-0.2135055921172181,0.5217015351387553,-0.3740955898135426,-0.293451019058316,-0.1150567575292664,-0.2253008239293049,-0.290010319948491,-0.3239821600350024,-0.1012891009326184,-0.1675250786147283,0.06108208546445218,2.216182443694473,-0.08052621561673007,1.57855569888051,-0.5682874884866064,-0.6163914173936342,-0.8444755298118581,0.08923703650749044,-0.1879094445190025,0.2449012070275818,-0.3513046252451474,-0.3436530442701502,-0.1416929525651272,-0.07505757645602092,-0.03177104938159429,-0.2650392720078891,-0.3850481610399743,-0.3358740172519027,0.4377359237791772,0.3415400233408136,-0.1823656792524332,0.03507877358832868,-1.7945986992472,-0.2620027474053839,0.2130412339027579,1.859217234127438,-0.3571115842277976,1.271931191837168,-0.418548513038478,-0.0962245457563683,-0.6972694470132947,-0.0327593513170966,0.08222929412963791,-0.5714717758702851,0.3946021487889708,0.7373616450571004,0.1687494027583686,-0.3330674559862684,0.02213365236830696,-0.1229695000952494,-0.2321161647070787,-0.3664374881091507,-0.02175374945891666,-1.783601143886463,0.1536094014516872,-0.453193046204993,-0.7448674277680674,-0.9428767452972346,0.1614165379118647,-0.5124978478371918,0.4044629570584219,0.3018566762695245,-0.4276503756706404,0.07424153305890641,-0.1749253618277065,-1.186764581365492,-0.2867822635389476,-0.1379744550222119,-0.3094432180936675,-0.1981772813330697,1.51953916759388,-0.3519212886679786,0.2575458169558099,-0.3821134342104967,0.6368650501708686,-0.07876527127412991,0.8304743005059202,-0.9171272545027912,-0.2747802275066704,-0.5479381072544014,0.1155160531749459,1.048696121714374,-0.8073222129957273,-0.3428872508475099,-1.014682207851111,-0.3550132536513019,0.9917647678404249,-1.424348047085731,0.006159865871857049,-0.5016358959524565,-0.2444044082327498,0.02037174506324971,-0.4036583239776257,0.619364923437106,-0.2215060578773421,-0.2969357199612686,0.9482286004191826,-0.6443220896878501,-0.5079091394550297,-0.4929310708561314,0.2710588944508919,-0.2875611582019947,0.07392748452622859,-0.2364404135883695,-0.4395691247828937,-0.2116376822396808,-0.08366016545239474,-0.05388233794599497,-0.06413249618594651,0.7752554697682622,0.7652961085257538,-0.2847082674518013,-0.4338372979095571,-1.236524186841217,0.227154348976558,-0.3559666411906896,-1.001691607979732,-0.2650947944574052,-0.1963334549174531,-0.271486110693988,0.309134907480503,-0.9953976773905975,0.639022499223965,0.5875938764778009,-0.1159632528185967,-0.1963935180139369,-0.02573470166475181,-0.5730733829725818,-0.432775330529561,-0.5950757625330302,-0.0462755253041931,-0.5315151536970073,-0.390284901417747,-1.205745276054694,0.4899673491387287,0.07581220647759948,0.1400548647942634,-0.04699964978816482,-0.05165811637674109,-0.2918446610896014,-0.8250490920799806,0.09377412885330377,1.270134744806583,-0.07010698970707865,0.2632657903313534,-0.2423203342099056,-0.5570835707649897,-0.1441639117368086,0.4401891217617589,-0.2181502759571516,-0.2746999946819056,-0.3528421448464267,-0.2261136767517991,-0.6500636868528479,0.161205508468499,-1.093879404439559,0.177230106911287,-0.004473684599704353,0.1331422979306299,0.08545496004152853,-0.312809706643799,0.8064512818980276,-1.363965910289379,-0.241163130967878,-0.5578373098713338,-0.3104186228050285,-0.9315319994004545,-0.4688475572326491,0.6615936831327014,-0.1754434056274398,-0.1353374508309356,-0.2455368454142663,-0.4057506446637592,0.1221417946861551,1.413887153472308,0.04293194608523349,-0.1816168554321769,-2.226696421656162,-0.2829647630146688,0.1818117869020255,-0.1539456754725279,-0.05008280057631986,1.2164977585559,0.5502424388404473,-1.019069739395977,-0.492958879941532,-1.140736531857863,-0.2427734439536452,-0.8688270082607167,-0.3329887722889247,0.6589065533992238,-0.8311125244271999,-0.5305185624708301,-0.2682716876286367,-0.4372783788658692,-0.02125318921134109,-0.1322940454616501,-0.0559684877484341,0.04215732465221168,-1.285240924783707,-0.3706205929561637,-0.03898073181562225,1.011472404194314,-0.4585752350767435,-0.270685627713296,-0.1901569742009406,-0.1209508675067272,1.130076504954552,0.4211601595192588,0.1477139225375531,0.04146521637791492,0.1862571980827699,-0.06057490009834589,-0.09082636885367978,0.3166955935089706,0.06638404306778585,-0.09637894701553669,0.004328938194729913,0.2679982748365526,-0.4706458610259953,0.07505876112643038,-0.1236016799168764,-0.1052509382782306,-0.9265235892858914,-0.5244478460547576,-0.6487247470352659,0.05308831899320864,0.03286541607249815,-0.06920873332446881,-0.5556771802968099,-0.9927714791408345,0.3516052734070805,-0.4499845102186726,-1.814492404838518,-0.1504081769842135,0.00807011817882192,-0.3114339377838545,-0.1750740505950042,-0.5303722103462237,-0.4597700522025695,-0.05734763254656575,-0.4549762653014108,-0.6335935652044671,-0.1858985759792924,-0.3140286202280359,0.3710797799285942,-0.390615771544202,-0.1714396119491783,-0.8270768592303379,-0.1150991733234598,0.2799768521425733,-0.3841997511971557,-0.4377831108854801,0.112974061129618,-0.6893742745119902,0.9313161485307249,0.1587618709721772,-0.1037283425707501,-0.3777947616355141,-0.09651084585713257,0.1335099176944057,-0.1794885809447192,0.2143978429964745,-0.09175236644072934,-0.1019274484489206,-0.4195323854098975,-1.508136556193768,-0.6554079013485157,-1.551271501558108,-0.6171716215447429,-0.07814009623998902,-0.5230784006410077,-0.4422039197423036,0.02251492290259712,0.3398717522300089,-0.2775172875618789,0.7336946097255453,0.02080439202168981,-0.3279519779411304,-0.2890096099286204,0.6122761479302468,-0.3495317989241228,-0.06815843179426706,-0.9375517471694841,-0.1120773938642555,0.1146200711280493,0.8589833001004323,-0.3653938757763279,-0.3074708229177834,-0.1759042432406177,0.1273074767007372,-0.0788522286472846,-0.4185661204660436,-0.06619343590672418,0.0110299126682588,-0.208972008307378,0.1123979620022501,-0.1357396958995056,-0.4772224267563215,-0.06999024579923602,0.2397044733253495,0.2133918184000796,0.5689066370422776,-0.1750375184575712,0.5722883815977746,-0.3471227813333203,0.3540398440518606,-0.5231075246808371,-0.04283598070677725,-0.3212908256967604,0.3120158624227917,-2.334176536284746,-0.1544199884534231,-0.2989544245899397,-0.09174133217537744,-0.4823988675218042,-0.4359829959335936,0.3409945325828282,-0.6297583179823989,-0.1902453012637856,0.1176194883893244,0.06718891179603113,0.5088938203411502,-0.6306213615285111,0.3329316505188843,-1.276680235812689,0.3801281524495995,0.1082837472220162,0.04240062714830373,-0.3350454508486767,0.1496170021214869,-0.1479233048841251,-0.03894170463348198,-0.4515671805902376,-0.07840614515774885,0.09349214548493837,-0.1945611590864614,-0.5495270639946884,-0.2809638903226835,-0.04606737846770405,-0.4039246148337821,0.5299024209104717,-0.8064831163311934,0.05187676637278833,0.6631214439779809,-0.224509764082911,0.8268760291855719,-0.6164748719354759,-0.07301521353851462,0.1163929227879835,0.6777089164374031,0.1677693982537648,0.1945680611927146,0.5068897263868076,0.2111396873068129,-0.2514525982108191,-0.5867870602337507,0.8425487882527727,0.08982657084242456,0.9865299796064162,-1.036957481820455,-0.4772975722401606,-0.1638108459119985,0.1729038455291643,1.164470618651685,-0.03016428526321721,-0.2010348042644999,-2.928263181463512,-0.1798542354362097,-0.9757802172585487,-0.422608353455634,-0.5617470642318078,-0.2684423572312052,0.1226655900884943,-0.2314522684038867,-0.1651757526409296,-0.3341448717746539,-0.1308945810900957,2.764141763082406,-0.3745010515387102,1.452016739395981,0.458370302303783,-0.456625456151977,-0.2162335205553904,0.05917215427436927,-0.1971214615120483,-1.202510328697253,-0.3499206827229303,0.6573878613701825,-0.4818624621846914,0.227536499326694,-0.6525269487494234,0.2288688635717374,-0.06882209351807834,-0.3130152786147747,-0.1094186975279338,-0.2792345185623907,0.2921980823029959,-1.131359735948788,-0.2982677897650022,-1.552161845214772,-1.078120314789195,-0.007598586440486372,0.2082681102262563,-0.04989302149249063,-0.2429844966435516,-1.107960635131737,-0.2365601474926632,-0.05557196985442178,-0.1500901422732646,0.3776752619941028,0.1385239918731974,-0.09076762305864283,-0.4231153586851845,-0.2832372311290023,-0.4126708298830437,0.1819989097943025,-0.371962356315052,-0.3706635048825159,0.0456432653286001,-0.8524506941871082,0.1201398193444336,0.2759270709003093,-0.2100201211349648,-0.224694968854223,0.3078387994704509,-0.03266309259131582,-0.3290683787896792,-0.5082922980957832,0.4654133521317178,-0.2617494049899285,0.2646557067558314,-0.1553615375302733,-0.3187269101374344,0.1171094503385982,0.1816748859657871,-0.4839781579374004,-0.3288282868412956,-0.4589933893102902,0.3049063947797526,-0.008390691349795542,-0.2632759049320919,-0.7521636499322915,-0.04928689728448531,-0.2476963532777453,-0.0202060422947779,0.1355137688826504,-0.04879224146310492,-0.07612387543494881,-0.4361227603853282,-0.04029119609824607,0.8321792913357308,0.3194528924697857,-0.2363322093319396,0.4195043614328945,-0.2772893551946293,-0.4319411002822685,-0.3009794118788513,0.1785264073439071,0.1945967604286202,0.2284333185376767,0.001329060955525071,-0.2898494226119491,-0.2914374037520374,-0.02005890259053417,-0.03586920645058501,-0.2276143543217315,-0.7648277330677122,0.006149737431053948,-1.45851504764132,0.3846132005565353,0.5414442057935125,-0.8961970086354317,-0.4352200998409141,-0.1387523029587526,-0.5370018478537286,-0.7699805534997787,0.05901795588639645,1.634178125123043,0.6621831828688297,-0.04328236341791462,-0.4847419435843198,-0.2126328017611679,-0.5228845795316335,-0.4206090047897111,-0.9596841068295759,0.5597924900548861,0.1016721659275056,-2.316642885226134,-0.0395464640580382,-0.0153695940568972,0.1003822080722991,0.2323927029755359,-0.2511869487404776,-0.5845353470844185,-0.2634195544536392,-1.2553491415216,0.5171745160177371,-0.8936247269411278,-1.248345159146716,-1.00544539909136,-0.6638058717908507,1.200515283292933,0.2044241716386965,-1.280201558449135,-0.4769220172432216,-0.03098951357832081,0.223934772873389,-0.2899369867133728,-0.2071004040262269,-0.7685294152594442,-1.328179151784706,0.1107011857039544,-0.5077758996324592,-0.8144549685992011,-0.6744279757768322,-0.5817522527873907,-1.195450163769497,-0.2202742723818787,-0.2584616730759839,0.1180985485898415,-0.5605832479170294,0.3272295332254209,-0.5845282887147993,-0.4994007113864711,-0.1549488337037175,-0.3371787761527851,0.2989745536611694,0.2345724732307549,0.4883668667528834,0.1404534666604252,0.2698996442527191,-0.03478501856708968,0.3212874958077917,0.5638647077667146,0.6033879747337603,-0.7007435445687991,0.5484181015822744,-0.4198332425631245,-1.378216736086121,-0.8515125683300993,0.1399408026188302,-0.3556542969554009,-0.1868685704568945,0.1042032984643939,0.09478864296982108,-0.468191843971587,-0.2777252756536919,0.2105287295190728,-0.9477264627789493,-0.1753254742241547,0.4088644519578382,0.1746396932555039,-0.6531743833449707,-0.5971846456562997,-0.4426460684977567,-0.4011921894575408,-0.6359897186065321,-0.1827495246574087,-0.03658341346659981,-0.8154888596772412,0.09727178709291162,-0.08068161183397947,-0.1452056006300245,-0.9519418054020389,0.1626772658435181,0.04552248170306828,-0.3097215347045341,-0.3083672358982459,-0.4057001434709115,-0.3013474939759888,0.03635473818076671,-0.738909596737865,-0.7055091867003881,-0.501662652060047,-0.2742284126888789,-0.05712451216317794,-0.3675138503899711,0.1027577259011431,-0.3699896526716132,0.04815131531245495,-0.3448744351066086,0.48969212993875,0.1200782767131984,0.02141941860486047,1.291354289523604,-0.1409590196529782,-0.1538341404849067,-0.4038493814353726,-0.2255901818606246,-0.1496571783294519,0.1521033510708693,0.2935224792684442,-0.04089924831727724,-1.152767222712167,-0.735885696678423,-0.3516980963485287,0.05660222043333368,-0.3435385271324646,-0.7945563807213527,-0.7593467371062244,0.4657743311305216,0.4162144263010031,-0.2986322097054345,0.105809182149331,-0.1044339999827333,-0.7970179922977604,0.04136855839939323,-0.5572786157507432,1.247330662282551,-0.312669096287779,0.4884404572257493,0.8718030338572936,-0.1721367629408861,-0.5870762170360484,-0.100852194897286,0.4881435710201341,0.7399432641004525,-0.9099051568132337,-0.3058364285153103,-0.6818527666891658,-0.4569872298433654,-0.3897785543949172,-1.345251087933347,-0.5375072950849609,0.8563392241314091,-0.5566716251210514,-0.3539351399008046,0.4948220950919268,-0.2619100866967308,-0.4204809025567997,-0.169849038310989,-0.6287782990917669,-0.4941531247150781,-0.3511067894123636,0.4374965703959285,-0.2047176256666038,-0.5806980356910295,-0.147410395792334,-0.09824527657907985,-0.02622111598118011,0.4508852187183363,-0.5623330069647884,1.480435363992873,0.2773500024673251,-0.1459668052324907,0.1031765301875432,-0.8040574791688704,-0.3681699939083793,-0.5009357691690111,-0.5554025235116385,-0.4863399219350469,-0.1676695606405129,-0.263318883876673,0.4807301585978888,0.860223165789245,0.2962349872578978,-0.1028243144015317,-1.03537072569526,0.04375321165417259,0.04377457025097391,0.758512381595204,-0.3519974738447683,0.0701748030151071,-0.2076469227498081,0.787007467727954,1.863992245441285,0.483412574093901,-0.2483430604487363,0.1849713135567726,0.1218726748186905,-0.08582324959236375,0.5339359057225881,-0.7314692618627526,-0.05533583837811359,0.3393582549949856,-0.1825859949290527,-0.3812516854302069,-0.5967247550217647,-1.359975384099251,-0.1084224311700751,-0.3674299787573636,-0.06641119740239869,-0.5396268768094664,-0.124525547273826,0.1515199479326358,0.09264976939080781,-0.2364742546991077,0.05297625056265227,0.2214946137202108,-0.8929327592759665,-0.4437476133715097,-0.01656480400483584,-0.4815005264244696,-0.1378715753291741,-0.5699617979015565,-1.718536178010639,-0.9830641679783202,0.2375098977568515,0.2104092767357968,1.239181808585495,-0.4215558347832665,0.3114775367610451,-0.08558648119436124,-0.6113849482622985,-0.5650324562522685,0.3518211094180256,0.2512022532071987,-0.9583116860699437,-0.7775709184998149,-0.9828740529891007,-1.398279176302406,0.5700371385916918,-1.249418459428842,-0.5996388946186652,-0.3244975737198578,-1.002722808153506,-0.1525489686184402,-0.3576949420232635,0.2826064751683001,0.8043224038074631,0.1704832150115946,-0.7708574035070225,-0.3857675988908102,-1.234769397979312,-1.206089890440125,1.861188228503854,-0.2943813240733681,2.239370764462606,0.6262193070521045,-1.432911131171851,-0.008506178946399859,-0.5012506838795867,-0.05051061356511817,0.1405750688338782,0.2414025118158322,0.07788408156319708,-0.1376325778260162,-0.036471426469662,0.3598142058149765,-0.9619503843533113,-0.3829647788836736,-0.5461014024348215,-0.271045313897141,-0.3276213969146178,-0.6281431588299787,1.51220420625407,0.4037404053775858,-1.125731148906882,0.4149770913663664,-0.4440469753774126,-0.2634846390063194,0.3542682898420528,-0.115301583771292,-0.1135051377662717,0.004742457939429908,0.218812144406991,-0.520110035939243,-0.3216993554723466,0.2421907867606928,0.4070100046376004,-0.2282780060808592,-0.1153622648862492,-0.2035499729977152,-0.1373747506672539,-0.4922311241076467,-1.365698844831527,-0.6823131480203702,-0.2602512701882023,1.117221817156181,-0.4228496667901024,0.3274447404499753,-0.006025407110397864,-0.479417682808464,-0.4311714858870169,0.05831229694342049,-0.2566677063601367,-0.0659631549960531,-0.7169731130856347,-0.8315395102322961,0.5559680655029108,0.6854163713106444,0.9143020002496614,-1.37043999774136,-0.06338482626359573,0.5427288484782702,0.4203626750943299,-0.1517895976911575,-1.356248644982817,-0.2393592711869964,0.339238541418806,-1.272508956576536,0.1395889519642609,0.3723925211909576,-1.404980537893157,0.03617332540883227,0.3845119585944538,0.6259057355051189,-1.450037380581448,0.1146751481894628,-0.4892141631457594,-0.2245787034092373,0.1478276225791374,1.739080000318467,-0.3462841115601982,-0.3935962682223915,-0.4168125914544426,-0.03851885819935021,-0.3205617358008455,-0.1605809703829793,-0.8980926617867503,-0.05607231922599254,-0.1229510772844534,0.06499484745922458,-0.03251989449807036,0.2196145387969165,-0.5935209691070323,-0.521546438792954,0.08674591894783741,-1.001643676111108,-0.4027730924009078,-1.057282001999346,-1.090300575139008,1.069980717774877,-1.182912198684465,-0.9035203138129433,-0.3918694390733095,0.1168394008904743,-1.172883430584544,0.3505346337156818,-0.4097437539913816,-0.6731872213912319,-0.4236439833552529,0.06418779162210426,-0.2403026625984408,-0.4633704458117007,-0.09900584217536752,-0.1021406025738527,0.1377864366065983,-0.1783818870254093,-0.7443788814215693,-0.6119538229984606,0.5209282018553401,-0.8298738198580494,-1.448506537232553,-0.7921789867685446,-0.9084764281963227,-0.6553154752718051,-0.7309056814315711,-0.2179078509398807,0.1788753449895752,0.2877494142539459,0.459282303339284,-0.4668156859910224,0.02561748238045585,-0.420594118434747,-0.6544177428522293,-0.117352292639257,-0.195422409424298,-1.411994205697774,0.6070072235573618,-0.1862326679553439,0.1835380768481842,0.1235290638056749,0.09331781642406792,-0.1840558424109934,-0.8389827039196724,-0.2210808263395905,-0.1488568558173388,-0.768946906161768,-0.6259651995265493,-1.137081187429708,-0.1291967727073736,-0.8435007362772373,0.2749237584945478,-0.00645970474206935,0.2541595864847024,-0.3658057893135159,-0.4990373202373545,-0.5098193114466449,-0.9708425546151733,-0.5488812508525069,-0.1046646394633243,0.1166879415430408,-0.8721409051276316,0.3212272350101101,0.210664669072521,-0.3883839555298629,1.92369039415399,0.00313580229389749,-0.9003444342169452,-0.4358345427593581,-0.01088407702129248,-0.1415955641061783,-0.6896710381714751,0.4839976194347538,-0.4535129595855656,-0.4677226067868974,0.001582883280595846,1.195336666881887,-0.6716500087137576,0.05006451473555149,0.5250565246123373,-0.4615196236620734,-1.280550008139027,-0.5194309945328104,-0.5749283294222833,-0.1400336965957525,-0.198426046724648,0.1876960983961842,-0.319742252969025,0.004391835116417148,0.5310148216609794,-0.6031619135626611,-0.4239184886141354,-0.4467929925125387,-0.09483842529168156,-0.1684437872786243,0.3166762127990503,0.001327871687179136,-0.09436857098107279,0.3222820925949053,-0.08440818785981302,0.4824726109712982,-0.2292404868021481,-0.670787131049785,-0.2753715940907046,-0.3168023085291931,-0.5278684307758514,-1.288459409959009,0.6537360346231919,0.299788022924361,-0.6607809785730172,-0.1578923928707445,0.2510439322894429,0.3834875860216568,0.2360854157609205,-0.4821582502668785,-0.155297724555981,-1.016550033411752,0.1464057852393695,-1.259345251477109,-0.01636483895282618,-0.7635582571544012,0.5252794922691709,-0.9080905300693269,-0.2298598693202666,0.7247340329317211,0.6211379891184338,0.004178797829968312,-0.1177818919936453,-0.03125645871036376,0.2043042587950977,0.04071874008700238,-0.3181302018280068,0.09380282518565876,-0.3441804395983735,-0.0808928195710551,-0.1049719176705664,-0.2040839694848105,-0.1139387640519365,1.067761301783006,0.342575969598286,0.3725819438699117,0.5983404349044497,-0.04971082525550114,-0.391433873550851,-0.08983527398114977,0.7761369864796546,-0.07033033895530443,0.8733506188877758,-0.3783350577496716,0.5020303317802544,-0.2278835487110594,-0.1843017153953587,-0.2874403667106235,0.9051360559284837,0.2413377212451809,-0.03615196695397694,-0.6304890967598363,-0.8406855709661134,-0.185232381521063,-0.2499545842027819,-0.02000294422032928,-0.2424127745443224,0.04424015983534083,1.801191284982088,-0.5256109167105816,0.06552929422107476,0.5172184193727525,-0.1131389003467374,-0.2844202577811986,-0.2878725542837652,0.02159010931985082,-0.3083128464724886,0.1372800864234549,-0.681067347364724,-1.658329060965221,-1.064593326696069,-0.4670693561015692,-1.025697918379376,-0.3705222644600376,-1.908063115043006,-0.4878541107448063,-0.3298859121056839,0.4248360516224644,-0.769298115370827,-0.08158731965492885,-0.3047005280818499,-0.3414773443932421,-0.5091621985690312,0.05073892550640325,-0.2406687525873674,-0.122274031890153,-0.07983651276380958,0.2969178032372644,-0.09330066980037358,-0.2338477078376682,0.8242398870590655,0.3921809337318725,0.9094118579375188,-1.000925826479184,0.1591612645092138,-0.1483162046303065,-0.8353608924935424,0.05680237791521294,-0.1321995191128215,0.4247789178869785,-1.202263919734179,-0.9430143076189021,-0.2699853601737423,-0.1982405933719474,-0.3861078787825252,-0.1876536757960044,0.7446513716569779,0.8277241990522705,0.6904990996858851,-0.8987621834711522,-0.254019031797826,-0.2797246926565958,-0.4785987062196822,-0.3848393445081498,-1.08834370123316,-0.07451122697883575,-0.7582818201086636,0.1919103767428268,0.4351039807436829,-0.2494868011919929,2.741826866063624,-0.8337997472573984,-0.03057042492525184,-0.2285012319730164,-0.1717888000401595,0.09357593559260412,-0.007760780120064716,0.9296428694203004,-1.65916830025933,0.1666714859669793,-0.0632845656265945,-0.8022289173510148,-0.6703569477867976,-0.2307227740789371,0.0945139836879617,-0.2672408565626215,-0.3342431062540586,-0.203140853776263,0.2557933662094868,1.549041681818223,0.6599934932467387,-0.239297249170875,-0.7011108622960057,-0.2700660625241124,-0.3145602928649548,-0.3605732110528835,-0.1480111593799454,-0.4177643300297592,-0.1906481981468908,0.2310429355849337,-0.2401170659788039,-0.2138870293733939,-0.5404254763687767,-0.03649017053073181,-0.1039814709248412,-0.4442650147608734,0.02192638589487136,-0.5106249993631545,-0.2499087853952039,1.754326405350882,0.1935247242451357,-0.30046099767902,0.31347251257081,-0.3263320982554785,0.05368353028834018,-0.008938634306269223,-0.1072125613901963,-0.06412134482561095,-0.2756790993697061,0.3123551225298402,0.02693862056460989,-0.5353945810967796,0.4106329410880288,-0.1518120325748387,0.2277025797227172,0.1250846571276028,0.3574342856883431,-0.2492337130556462,0.3158108224264549,0.2580876298897343,-0.3130184634492014,-0.1606228419860236,0.540547979366655,0.4610590013161818,0.4578187068434972,0.00392683435647704,-0.4221249532111833,-0.01657341168654382,-0.4723136031019786,-0.1794553865943659,-0.1610809944146057,-0.3699359632068988,0.702525431676404,-0.288468205875634,-0.02052378647501402,0.2025613981523449,0.009062015267954823,0.5066114344821152,0.6050762884649125,0.2172727840199018,-0.1353481247264303,0.09209445717550356,-0.8991748631247825,-0.4298528097238764,1.907915881381575,0.1025130866133167,0.4638506371059838,-0.006407099507970023,-0.3145840368426437,0.3798832136861733,-0.987164815770423,-0.3785100148350039,-0.8247547557513242,-0.1303242035655535,-0.307916677602266,-0.1713128955471444,-0.3903950676217765,-0.2865869655688802,-0.5090704411429346,-0.4277212949889349,-0.5114067301203973,0.0385927711608899,-0.2834657052157045,-0.5230585720571511,0.8413370990624414,-0.3895552962912004,0.03238976093846419,-0.4946708228353161,-0.9405093283246463,0.1996356513361044,-0.2954178925204808,0.5771700166324523,-0.2631812340811046,-0.2354086032918411,-0.3409041851272544,-0.8446451677537965,-0.3203504302879752,0.1520936959148287,-0.993758533148086,-0.04852435718620065,-0.5955218427562166,-0.8182717988474129,0.1040816112764635,0.06432685383122415,-1.110754678454,-0.2838921998095296,-0.2875693379202733,-0.4132816024493817,0.2338668349158567,0.1303180913064949,0.1141244615435927,-0.1075802364758295,0.4529753304309899,0.4595516692644457,-0.1247753451357505,-0.9490789403556307,0.3442011680262134,-0.1578647057503843,2.43794990702795,0.003312892606811738,-0.00535322616356817,-0.02113295355835999,0.061703829157229,-0.7339889664847933,0.4758100128468786,0.2297898956378074,-0.2564803916363372,0.360043310860278,-0.9783309021899078,-0.06030126667181673,-0.1454195387021539,-0.138212447066646,-0.1816369055749681,0.2287831626197479,-0.3770896622263923,-0.07064913058598588,-0.797453825425717,-0.09616779443139946,-0.3274269145535434,0.1436488067426686,-0.06487422405546694,-0.01688259341257968,-0.8487179174291762,-0.970477433565113,-0.5598037018255257,-0.236720669829608,0.890389683893886,0.2012412947174174,-0.1227906995888244,0.2507922507270666,-0.5501952282713372,-0.1361794650482027,1.025294085124518,0.4560578979080854,-0.125177084341848,-0.2957149587258989,-0.3562343563875703,-0.7550421156865068,-0.22508799516633,-0.1066389785175876,0.1952041908878891,0.2802494623044414,-0.7531936150933285,0.4612825902830612,0.8588928161584356,0.04185188877509696,-0.5961847158470003,-1.220187171656826,-0.5574123986816198,0.5843983022314899,-0.009219488527385175,-0.2802723224604453,-0.444014459366169,-0.7558083525647268,-0.8659953837506105,-0.557019061043027,-0.09072537047207435,0.4518138915063059,0.03298134929767198,0.2947297900097119,-0.8556808942686425,-0.4424683334214076,0.127895153055602,0.1426389302343066,0.2426292946386162,-0.5612078523726313,-0.7069563620654534,0.6172093971397288,-0.8325484607571012,0.3572064211078155,0.05779890297465429,-0.01828632066201796,0.1848020858342093,-0.6738391739632527,-0.5976757998982559,-0.2550707778049314,-0.4361826573702592,-0.4606000821385047,-0.2127238502314412,0.3875704836659877,0.075162532081072,0.03052441606281273,-1.151364911120538,0.06873381194265479,-0.5925597903095903,-0.07642475829478075,0.2643213138282406,-0.2101316977406946,-0.2985903088186566,-0.3102615691002634,0.5542286173387792,0.381768840057014,-0.002584384749034167,-1.10428589670625,-0.7720284969646526,-0.8602113465207596,-1.183536254126605,0.9131032449839663,-0.1298325682160133,-0.8836761387540278,0.1641595836050402,-0.0107012375331962,-0.05783551415388166,0.0115557702147474,-0.3506405917223895,0.3681636636192333,-0.4467650728330257,-0.3523745458238261,0.09787802980106451,-0.0833313886686316,-0.2528569904859678,-0.08253523362387861,0.03975849888716156,2.033817157611076,-0.4954752796784094,-0.4089931723452887,0.5579100345511105,-0.3943065512864671,0.2587351118485235,-0.1525185957665839,0.3585240178466447,0.3625144950711739,-0.03571011552055849,0.1268041489463941,0.3625219001088713,-0.9899666284876006,-0.3152507294636063,-0.4505194467808646,0.5527536542233186,-0.6002384147552972,-0.3657092742315495,-0.9008650843694944,0.3503418370027618,-0.2569168546851346,0.04212421836796721,1.408332429644164,0.2465332097963611,0.7412397520228168,-0.1802815612343462,-0.1883850096349509,-1.1648900092023,0.2915631153340087,0.110500029100814,0.5425396755390743,-0.5571769749873933,-0.2404834122874347,0.3129799785970862,-0.6808629435948892,-0.1901793920115394,-0.4072244048896292,-0.04462589466800435,-0.2290303706263863,0.6768193238660201,0.007991095248126554,-0.1025122322684614,-0.2853928652580491,0.3577179148463022,0.1970299728091495,0.1646196177997606,-0.9750318056786533,-0.04280527489481872,0.9781740630923419,-0.3339642487474929,0.4487760294579565,0.187260859225044,-0.3501990917626058,-0.1241005565047698,-0.9097731779665587,-0.5060111428292799,0.07023528858971849,0.847704620559809,-0.528331470791269,0.01897849439457931,-0.3282198142000157,0.3550353052385717,0.4004354155194134,0.5959497063282404,0.6868103815642987,-0.2093757293962709,0.6650165457272419,0.5295198749592329,0.4619953947272988,0.5102560201283429,-0.1771014667838387,0.3166562381384207,-0.4708057374669729,-0.2001963209731281,-0.6475870805000479,0.08024049757312926,-0.2436750339128161,-0.6416351057191387,-0.009816200736994102,0.2806436784454547,0.6673159778862835,1.301671851018407,0.1284667719453199,-0.4648043870327682,-0.1522206183572756,-0.2262715968922346,-0.1785260011238791,0.302731463657653,-0.3543962405569425,-0.003625825036569238,0.4548649076195472,-0.1366876515480903,-0.8033923758632541,-0.07096569536884295,-0.1588176296249628,-0.3913356991302454,0.3623167805128671,-0.2164268653614496,-0.0165239836502789,-0.8422684206825873,0.06960055614865862,-0.6064709377149312,-0.7997643460757232,0.0295299993693583,-1.329009969360484,-0.9231735472572946,-0.9116564243652138,0.415776390175539,0.5838254260537106,0.8417568530815925,-0.7648976225389995,-0.9433907917796382,0.9394343586333517,-0.8545638864427103,-0.3530924349905367,0.7752466761952894,0.3060875922256816,0.2926883561854997,-0.02789473538565766,-0.02488618263247228,0.09251524450050404,0.01638116423652753,-0.9084498381029755,-0.06263415264253519,0.1555019370897112,-0.08831219186478784,-0.03610533581112996,0.3345197391713731,0.4310869939902281,-0.3015588176658338,1.067552424444409,0.1852358386816771,-0.2013254269306261,-0.314441896148022,-0.5417347803336416,-0.3569670004966864,-0.0777636638921852,0.06139992220164434,-0.03440993975346876,-0.1117576953289239,0.2058961743424493,-0.4213735868970881,0.5321919269279761,-0.1440555693627915,-0.2019220599824507,-0.009797139318503432,-0.4812609010191545,-0.5995600307492335,-0.2247131258535794,0.220554690327266,-0.5344676929078707,-0.4138145898451395,-0.4305055901683591,1.395069894979947,-1.149901593341604,-0.4213379261149188,0.0779400355712996,-0.1638284309320346,-0.43239894413333,0.02916185543689769,0.4836901313062759,0.4946556492309893,-0.2890999729420071,-0.06363840395405032,0.2780319684519454,0.1460064903125551,0.4585135435445566,0.1476559385658718,-0.04855970875468394,0.181973587275404,0.6601165865498928,0.09345298714567959,0.2492233626215451,-0.08509461871151676,-0.611566251651302,0.03797959125666659,1.143811242272388,0.6518244767811007,-0.4023509973316935,0.7479489007240536,-0.528794873604406,0.1272656455284319,0.2028126745962692,-0.2483811816075101,-0.770310730228747,-0.1705074916761999,-0.002422365568286319,0.1741730595309613,-0.2265316827258597,-0.311108419930122,-0.41460252479617,-0.2450296082976442,1.56515399589683,0.02679587000730698,-1.171970666090789,-0.168869473983493,0.3042908546587531,-0.3850407311462635,-0.9367859668701033,0.1902340720050879,-0.7406213798200202,0.139186811802537,0.1928328536628281,-0.2643787699307402,0.0199494934928397,-0.3013347062671979,-0.2418714929648322,0.04414102817398231,-1.222744105321967,0.1459787552384188,0.08214053551062929,-0.2329095228261422,0.1953362001220631,0.2092032900505634,-0.07301265427387764,0.3650900735460658,-0.4544379548515096,-0.7706950211253368,0.1109577753003275,0.1313878281788488,0.9359722970990662,-0.4424778368422738,-0.04411506104183253,0.7653074423637424,-0.01399592886696824,0.1226616237562527,0.2016724303002255,-0.06849468146582562,-0.520686098820031,0.9590601386914397,0.3731955303344602,7.678181018079334e-05,1.619332280363736,0.3695804095096627,0.1258253572066668,0.03575283528399874,0.007548697619234246,-0.07163417244984152,0.2142737493211543,0.16205236555833,0.04141892089506767,-0.3580892335547513,2.139512076044907,0.3916998623505785,0.04764216001927088,-0.1477659906510372,-0.07370551807697348,0.009843589924690815,-0.3928331956318979,-0.8201225034272461,-0.3262302055677054,-0.5313978926360243,-0.1990423368116017,0.2131148180155301,-0.4063823754131614,1.255569290514855,0.01287301272860926,0.7079712579803062,-0.097822503409438,0.05641584929092477,1.276841497095443,-0.1360857358544712,0.5273798573020539,-0.4564087981625896,-0.1921018591765719,-0.1629427319931512,0.316550452377472,-0.3868915591897147,0.17287888015036,0.2286917542207449,0.7888544847970725,0.513423092010114,0.6506065269877424,-0.2740337247125104,-0.3334503507570993,0.1639885400383788,-0.2026068156552316,-0.4836510846128862,-0.1322069269697383,-1.636426086420566,-0.106360374587615,0.6225432542675209,0.3646620470743822,-0.38198243904124,-0.3675936030384391,0.02219481657181189,-0.06002500322279324,-0.2190372500151162,0.1831576694804391,-0.3721365456414136,-0.7497794844193706,0.3726546530470667,-0.6799573069851342,1.689405557042803,-0.02890320989884086,-1.010320219006786,-0.6015293708286406,-0.6760182600225511,0.2692570827002825,-0.2475325776711925,1.478495142343946,-0.173677177784589,-0.6389570333304466,-0.04828033124230083,-0.3483124531797031,-0.1950850225778,-1.178161745949065,0.3435438969018372,-0.4222406311026062,0.5088430955770308,0.009916237306268843,0.7615942879150548,0.6009753798008661,-0.1473645425210303,0.2138313253597935,-0.8852674240140832,0.08314515165511782,-0.1158517223199422,0.1566511917087695,-0.2344467071867673,-0.1237713899468403,0.04671683564815733,-0.2181614326324922,-0.1948738338192823,0.2825560860812125,-0.7280535387746327,0.864492172016933,0.05932186860942389,0.08556888114318735,-0.1406532257183402,0.5034808513819609,-0.04279621545311833,0.4764233468854395,-0.5915170142943232,-0.07105644191569216,-1.350127340598741,-0.6930716210692199,-0.1036928660656513,0.3137880723141759,-0.04701890478941151,0.8178520444649852,-0.2175875234085956,-0.06187277258360539,0.4679236392903632,-0.3930860618855528,-0.3631751791039113,-0.9302860475624766,-0.4452067938407672,-0.5165173073816622,-0.8199399152730799,0.0398462848215726,0.1962272338890199,-1.473189638794082,0.3005533972441007,0.1238175027860897,0.1194755486660957,0.7424082508224107,-0.2878266501186107,0.1432482925759692,-0.5997329385136516,-0.02016131746517446,-0.04770737694419018,-0.0895494965643208,-0.8103490488462706,-0.0360887742712144,-0.2260847497475285,-0.3544585949356326,-0.6858186520968546,-0.3594724042543385,0.08093901108879337,0.6067445779756288,-0.008170289849114788,-0.2277679986049423,0.3510827071471475,-0.03954430878319173,-0.02287511004597658,-0.4600077065041826,1.562764324616816,-0.08812741367105552,0.07795155673393335,0.5527102048765525,-0.3704750473329871,0.1890980989876649,-0.9545600165098788,0.8621911109448772,0.432913388615657,-0.5292046282273444,-1.895044662999469,-0.02264850063853425,-0.3196473148340132,-0.3742314708707832,-0.1901560935854386,-0.3537515565986685,-0.7028406438865988,-0.6227639257817593,0.5608168154477975,-0.027456359473223,-0.3240324316739375,0.3851791571495922,-0.1041659493999114,-0.7043992037801161,0.3580051559805494,-0.8045348799165004,0.4748464441178282,0.7217655984630509,-0.5171827321897303,0.4891942621321852,0.3528010574216673,-0.4137067102045438,0.1554874394948732,-0.1797371416609024,-0.2772302404334442,-0.5009917867665726,-0.1707503001605133,0.1844568302558299,-1.003677428198065,0.6150646949240478,0.2396168186531578,-0.582272660905843,-0.2778288459596999,0.3657488469113066,0.6473351118330049,1.578045908322543,-0.0422918971681309,1.145992256955446,-0.1245581088352712,-0.4480432219929635,-0.2990064123995833,-0.2768194171405171,-1.053296804760886,-0.2828546261270903,0.5155996146594399,0.0717321047158434,-0.6730599440602073,-0.09941744000585963,-0.02124338743872618,-0.4898857702776084,-1.10116794620134,0.2410149960836291,0.764418236076055,-1.354949744073821,-0.5075360211158456,-0.6023222839600648,-0.2268098645251443,-0.1877458977005485,-0.1439448471800771,-0.2231668789610559,0.1102964884445507,-0.4052693567388418,-0.4659282353550939,-1.181937563911391,-0.366026270113154,-1.006153501679799,0.6115443473850182,0.713001332630976,0.2561028265471418,0.272586859718113,-0.1196157479226632,-0.9813756904276494,1.501876849615102,0.02016417927174079,2.205716957948496,-0.4322845565876089,1.354486975274454,-0.8095068065898922,-0.4948090530496936,0.2493375463473553,0.117619738599436,0.4937037929715682,0.9804024170135709,0.002079050236408268,-0.2029305122542999,-0.1209099369834225,-0.4930280510100202,0.6991237338877642,-0.04746754177665255,0.1248096611136453,-0.5097739353594907,1.020191563443348,1.169337938164077,1.233325316551034,-1.306157798642516,-1.052631597919781,-0.2768731168469693,-0.6501303387996954,1.404345309744932,1.122906328705177,0.02635307315606375,0.8846309769304997,-0.7855102563632367,0.9819498659612261,-1.179120590874628,-0.2377415181110471,-0.6554413265091199,-0.7138939424769056,0.1213494571680777,-0.8956975294158451,-0.175980188360653,0.0958893726640246,-0.4647286604205376,-0.0570501199586195,-0.08371977890760308,-1.727365683372419,-0.3148199270308713,-0.205753417479128,-0.3671358506376248,0.3894693657666179,-0.08125615394942599,-0.3917067330035641,-0.05938934467988031,-0.4310219897435452,0.05011212814877575,0.4016235287672389,0.09194120491948592,-0.3335644647781186,-0.4812477416729278,0.04654974656421994,-0.2887237714497563,0.6444433542812753,-0.3837439233560622,-0.1152486732029699,-0.7325196481626507,-0.06460994837222518,-0.564263245447421,-0.2712551245597014,-0.3715899413506777,-0.4591264656890483,0.1880881908834456,-0.800580139874328,-1.091870130013943,-0.4395794780816548,0.1117439812833124,-1.165917701695302,0.08516900326904218,0.9098625606305333,0.09333430977881825,-0.4630099654504095,-0.05065230833751953,-0.7378830907694575,0.02074990323670291,-0.184336548874494,-0.2075232592868621,-0.5402734328415919,-0.6348825635458267,-1.077267884813537,-1.084486403909412,0.05866721503605452,-1.083759856612369,-0.3687679347271301,-1.246416451541792,-0.6550885664628999,-0.1833973284667326,-0.3758454502069263,0.06786697947438672,-0.6873382091059933,0.07515284523165053,-0.1985604867376218,-0.5495317902850941,0.7124949202406343,1.232194794666292,-0.7919914823532495,-0.3592132190212222,1.47486162424945,0.6122033035560038,-0.3471941098677521,0.2078954036696389,-0.193356466074857,0.1437652879016171,-0.01195467597561799,0.1366578737176343,-0.03704797014034683,-0.5115079746638724,0.810016129049424,0.1938354021337711,0.8550540808291509,-0.2792899341665289,1.631521941778641,0.8461038661497415,0.7813690443214276,0.533152072174109,-0.08263275757556074,-0.08859701984965675,0.07728750771610947,-0.3587819062046783,-1.08894177317709,-0.2607810041419334,-0.5474055030558234,-0.4741160421939236,0.2131116615996306,0.1745195866449326,-0.07104954777768961,0.4545369688588236,0.2444832132220707,-0.5550869819533901,-0.1659275352172404,-0.5881556189068636,-0.5548251187005196,-0.164438617072374,-0.3718905928979937,0.01450190667314201,-0.500784598515676,-0.2158188552140855,-0.8320262324379563,1.261520211538366,-0.3462827159801092,0.2089820155858007,0.6459946337461802,-0.7032887156360823,0.6033647125229434,-0.3561476811042532,-0.1963614379976309,-0.2607320710502128,-0.3192018097706273,-0.1821527790259819,0.3027202026778961,0.1985997507956498,-0.5954950630690975,-0.4594704157035573,-0.5226514005622592,-0.9672166506215562,-0.1983700865309214,-0.1089925391180162,0.07589801010971846,-0.6058244313457223,0.2006713064889361,-0.207230664193364,0.6858138728399884,0.4814054138584907,-0.1008961792741202,0.2302478256123949,0.02590457037918949,-0.206127451231371,-0.6639250473652493,-0.9141377930247766,1.378033757932404,0.3360788471494365,1.440529810910115,0.1561075232055743,0.1149796383467404,0.08556541689830825,-0.3758951166115866,-0.5599496512990955,-0.3353414412598019,-0.00894822168913036,-0.3646354030667492,-0.2389756476536085,-0.2646394515301043,-0.3721619188828073,0.2165976752174126,-0.1774534992655169,-0.6690888364139727,-0.1357630418468926,-0.08470507400866294,0.8540575445251964,1.342772111977366,1.160288494351243,1.331738284852272,-1.119952614467533,-0.04544727667543578,-0.2992463684124755,-0.8342393650103321,-0.9403070576895979,2.022129571238914,2.184595806316408,0.366063766154314,0.06498666982189921,-0.1992267873427085,-0.4114765613871629,-0.08904042884236682,-0.5064683769294341,-0.836462824533306,-0.2679553970486753,-0.7572278762664065,0.390581544987,-0.3030598020950681,-0.1163098270483445,-0.1906627030151268,-0.1147144072129938,-0.6620619329059716,-0.4367186834984297,0.4731308487420341,0.004233578487470353,-0.8256072576789475,-0.4429480626627914,0.9438543642971824,-1.26403788169492,0.4463718000977606,0.09069007539011305,0.8381446117891552,0.469674301207203,1.16159428339135,-0.1558628516425891,-0.5255425739293903,-0.3719281110811681,-0.001546608594962505,-0.2367232238555622,-0.06453735806520969,-1.169053654284838,0.4013901741509879,-0.09494454394231897,-0.1784642753500281,-0.2440749932425014,-0.3915904168117931,-0.7072882137806626,0.1104178956350335,-0.1208685486151611,0.2239713139680033,-0.2474918823704393,0.5641526368486615,-0.4358296193402615,-0.6120631328496755,0.2309440139863979,-0.1487650925569712,1.035188608272855,-0.06635128469808013,0.3302691745373866,0.6409294888046358,-0.2606295882217204,-0.07604799032768099,-0.3906775183871813,-0.03801584805417925,-0.155755203508822,0.2624051993585386,-0.03494289330849461,0.0670068654839764,-0.3748768201146728,-0.2896585966452007,1.434876915571451,0.1687854190677629,-0.3652250807074442,-0.2014046875184207,0.07283411161423077,-0.2519977438027541,-0.8574444704045837,-0.2431971403021715,1.508517771145543,-0.1173991138828833,-0.3692067973611216,0.5497135432337992,1.304962603327193,0.004242769633511868,0.04846334124980693,-0.2075913943128923,0.08391283171682322,0.3317088447855571,0.2741269221740771,0.7153395146789877,-0.4601185387675034,0.05630557986273345,-0.9178787346027285,-0.6072721682337794,0.3405050714607379,-0.9684409478958935,-0.3065952267106985,-0.1160110183623758,-0.2213624817891371,0.02314278315762931,-0.6111178775520417,0.4796201676590059,0.05275565640556134,0.002475948574105443,-0.01120957479742777,0.3933371628649228,-0.04460205702997043,-0.2144262373893854,-0.1795701964466948,-0.1210524025377936,-0.2878348870677439,-0.3520498722689638,0.3223899055660612,0.01534937037550104,0.0406063951567778,1.507286163433621,0.2818442599854797,-0.201572302682361,0.00980517481190139,-0.6148022794555693,-0.01222123054141412,0.2865066204456632,-0.898877078415238,0.2293240359323133,0.05354140081673361,0.0657561581370812,-0.7337623585517193,-0.3437783431096786,-0.4201792655713872,-0.4448926825811026,-0.2561460667423816,0.3393093118863815,0.1874797429639253,0.07954652261271566,-0.4131220961590586,0.4388905749881868,-0.3873872667027987,-0.4606540887672134,0.007493845058804554,-0.06725895635728854,0.3452474188697596,-0.6491168778542946,-0.08142408657032524,0.2571202205054365,0.8987084299854476,-0.3755887523864081,-0.8782910152743981,-0.1218218836846234,-0.2973969892448037,-0.2594795463293171,-0.9067348667026554,-0.05809865910547474,0.04718776592536194,0.03288935744398438,-0.2273164984667236,0.008936311721287284,0.002519225954263949,-0.004727824731715508,0.2139503368546158,0.07449464669223092,0.7029153537863646,0.1335086464226307,-0.1959911393784232,-0.6029904864653272,-0.837330988022523,0.4867372660033427,-0.2988128672655211,0.07912718383516301,-0.2215144605465081,0.4283698271132643,-0.9643813048065081,-0.2430055449794243,-0.4018367430649685,-0.7328726648027846,-0.4571846531677465,-0.492755023176899,0.740484691977305,0.1494794235369189,0.458887671707859,-0.5514746736190792,0.182867732689272,-0.196278367188311,0.1216838211836074,-0.202387332662615,-0.2783193403999792,0.08781535165230717,-0.5622271554981538,-0.2514568458582941,-0.4522345264514408,-0.6672921797157804,-0.5858658618409921,-0.9493333649478887,-0.3676666930014983,0.3667782678431569,-0.7447789628370446,-1.088170952023848,-0.2913606549021486,-0.6433801964697747,-0.06427573081141398,-0.7569786070067049,-0.3109147461307908,-0.5052879155021288,0.5148412813964287,-0.1095765082339019,0.08619301329808321,-0.8672287209630593,0.6606280200407803,-1.410256021993586,-0.906718751436141,-0.04492781085037626,0.06158465455146494,-0.1611807753300571,-0.6707158643106585,-0.5939143772420372,-0.3677445135813366,0.01160589558545686,1.287972751567051,0.01427484898494056,0.4683830960814946,-0.5012484297461784,0.1851766350155776,-0.06593509939845904,0.0203613814196763,-0.02130745414560546,0.1089253020278995,0.2787765990331627,0.2936796626108403,-0.1117474473208622,0.1769794353580363,0.497287443059492,-0.3510392148227043,0.1045399193523866,-0.4151850538671061,0.2857487831837734,-0.03086810424085798,-0.02442885531907938,-0.697689341691646,0.01921006835598746,-0.5391469709656419,0.5109371034530404,-0.6371886855579472,-0.3497391192322628,-0.6850742317113203,-0.3095741251920083,-0.1932200401008302,-0.6942628000386687,0.09048923429329653,-0.09002221944279243,-0.05939427166801294,-0.3030611239061249,-0.4403629654328366,0.08858217038785025,-0.1460412400612859,-0.4272366619738049,-0.6785059686681739,1.74017632195268,-0.2687077467731516,-1.014636406973,0.06806052097168838,0.8037847747802748,-0.2175527684470664,0.233086084087718,0.4249034885911513,0.2415598883990636,-0.5827591382328119,0.05339330877053691,0.2716396338865329,-0.6176401308214025,0.2165838081240534,-0.8241635131327599,0.4046092855781315,0.117249832347619,0.9997375796604175,-0.5542842521942998,-0.404924794367179,-0.7169245222300102,-0.4382585555151739,0.07714820523168278,-0.286257305771535,1.652383722776019,0.2569028912773943,-0.2211557333002507,0.3611810591635653,-0.4393374567675263,-0.3592847762005476,0.2684294485374085,-0.0169272615693381,-0.9890687399273838,-0.1903107549929812,-0.02300480206691916,-0.9597162720280473,-0.7301684327025866,-0.4232714720011072,-0.3842106423501437,-0.8410913006546191,-0.04783243272397757,-0.1155757207286749,-1.134624901732221,-1.07285094743334,-1.182744496201288,0.1866082771760183,0.2010935306812881,1.751171279618873,-0.3510160671414703,-0.3255761321344714,-0.493061711902315,0.5486616317187616,0.2560913380579035,0.5991747657640604,0.1669368944210573,0.4196721433834927,-0.96075999759552,-0.6010283406412203,-0.6180750830506712,0.06608286256845762,-0.4626347317735105,-0.4002805197069836,-0.804091348202421,-0.5085597155917139,-0.3384375999194983,-0.3032496506830661,-0.2527261061051475,-0.5404642042370148,-0.4547826962918494,-0.857332821176062,1.12135031066452,0.9398067934107031,-0.7011531675254267,-0.5152013675324418,-0.4023428420456358,-0.3654411119384463,0.2327261487279811,-0.1958705748115421,-0.1431705504586223,-0.6130108904612398,-0.4107416412321189,-0.2781498173626692,0.08309835905378908,-0.3351004599946059,0.1210595027685962,-0.5184072928076037,-0.448102906725631,-0.4703365246081017,-0.01599603816787329,-0.5498476782090722,0.1132061945924244,-0.3459448816482388,-0.7764974623133609,-0.04417124921641361,-0.2185122624708997,-0.5166451491603056,-0.1744631249503843,-0.3450557635839388,-0.377205438546786,0.2614953374053063,-0.993816464724416,0.05073752107764332,-0.2439700290948164,0.03991381270629327,-1.32325600886247,-0.339564980073182,-0.1817475254431404,-0.3141828636172701,-0.1020018504937424,-0.3808608987121601,-0.3042435033114196,0.3211909470289238,0.307493032430235,1.87921191637653e-05,0.7575629525601799,0.6120856166112375,-0.2772565225422821,-0.248206357350443,-0.8953045742305513,-0.9748445408270446,-0.1208725481312098,-0.665164402245319,-0.6960631616657196,-0.1288935519030626,-0.9224836917797352,-0.1776900081801713,-0.3644825188451783,-0.2384707969408845,-0.8559955279433972,-0.578987792586895,-0.02900774743171718,-0.09298917435038438,-0.9280838350354829,-0.5021023629982073,-0.6289482974989565,0.1803583472455075,-0.634704350916423,-0.06037912724335348,-1.056861945464214,1.56983374586228,-0.1242027947487831,-1.074209924213062,-0.3583185549327728,0.4905195316002041,-0.8597191503929728,-0.005720902053460777,0.2688919305992676,0.0890227642303304,-1.3364702471142,-0.05932019760937206,-0.7782092449978549,-0.3628429916123955,-0.3701588347200541,0.3967353997230455,-0.7432078540412463,-0.1286557389904274,1.203344656816145,0.1056634509524675,0.6789077809998826,-0.3144569610625013,-0.6942614471047075,-0.3316647986936994,-0.1728118937521184,-0.341723086949858,-0.009413092005080502,-0.4329311583272305,-0.2100714341797255,0.129357561434081,0.01108388679654539,0.0483820319157292,-1.05343931127705,0.07957437585760313,-0.3975014166541488,-0.4004333323178537,-0.7717179229891965,-0.3826193574999759,0.5655372486328144,-0.1329361321679783,-0.6683000063215719,-0.1185206300994898,0.06485773647686155,0.2313865666901329,-0.2487201929035115,-0.1118188502429381,-0.3007376434084188,0.2112386117241514,0.1763924141057469,-0.7813119414489709,-2.069285868920613,-1.05413299841013,-0.2412053233022065,0.5720689807515503,-0.6222632851217603,-0.1698154031664909,-0.0471024055875158,1.123893625760882,-0.1201265254019231,-0.02071840645473614,0.2191575138599911,0.03708998325663182,-0.002242082657664899,-0.5663252327575581,-0.2410836251005688,-0.2160717475164269,-0.5777350819346989,-1.037467701656105,0.06019843149936692,-0.007319992109197211,0.2348084427267333,-0.8553494927609847,-0.4784593401388955,-0.8078410828726907,-0.5407470090428601,-0.9040241942592409,0.3448578831235212,-0.5580951057398196,-0.4627829461377884,-0.1463892866696896,-0.290422426184679,-0.2133207675842092,-0.4720238550392117,-0.135897944201343,0.1515515720584249,0.001574992845145562,-0.1923464781351487,-0.4317162455878954,-0.4342967224603921,-0.1191747620942801,0.1847175759639217,-0.463126757113736,-0.4465644368319134,0.07551254577740681,-0.3394460372800503,-0.6248482948826842,-0.2525230784577853,0.4233586240487595,-0.4366220070186898,0.8929023200370352,-0.144746018279635,-0.03128446624381381,-1.206641542112565,-0.8911011144440533,-0.1887833545414909,0.0346037181819082,-0.1724007770514663,0.1384305551470566,-0.1836731990323528,-0.3310659829338454,-1.18823877432714,-0.1675938862089054,0.3517642422945705,-0.4183464416708981,-0.5857261977461226,0.4429250687983456,0.1804047174093073,-0.08026386253125632,0.2678113651388114,-0.0795542599542268,-0.1400894139480972,1.038439014720709,0.02255733156570813,-0.5049451350590074,-0.2815399675964185,0.03152098955368156,-0.1312304806536411,-0.8964641496405457,-1.602004174295564,-0.2414036629174068,-0.9839286834056152,-0.07402058497488126,-1.095522185111561,-0.362782386218991,0.09112858210825346,0.2316194312637702,0.3967676890444514,-0.6381633756674849,0.1327659677749428,-0.1583507448453216,-0.06898910978725373,-0.3064354519972525,-0.3999906782694103,0.08571634883052308,-0.3357953709064566,0.3238325265749905,-0.2098745601447704,-0.1394756441608055,-0.7801761098517119,-0.9096808430512474,-0.4630744917275742,-0.2134438164354509,-0.07940738018684225,-0.1965087938556521,1.279272785163904,-0.4880631466886107,-0.07520624704962718,-0.2966268396549172,-0.703130618068786,-0.02262345590802916,0.379429660782912,1.185924698738301,0.6436039796024828,0.01887051177574207,-0.008748135214712365,0.7747029301875348,0.1662861893581749,-1.159258625523695,-0.4972598806111566,0.6309985975783847,-0.3790261870509851,-0.402236737818797,0.285557358062995,-0.7060567684957315,-1.012265194693418,-0.1680095808273085,-1.011778142834366,0.1632101024678686,-0.07922700226281383,1.17481403174084,0.01592515532439321,0.08293418251615881,-0.3845680376449517,-0.1281039210295262,-0.2820918895452836,-0.1561237929750106,-0.3946068132066354,-0.20157529184769,0.07310891689709641,-0.6876339111515485,-0.376443110870828,-0.2462034242448837,-0.6009041712509003,-0.01847636747442251,-0.9463806453482974,-1.929929795129401,-0.2903135377004222,0.9217751913346591,0.04253415641571951,0.3534742845317914,-0.4008287095589198,-0.01796126237020092,-0.3162928244501031,-0.2083654304787444,-0.427194830408441,-0.2492614446500851,-0.1736060147924759,-0.05115426885981015,-0.2839185253304299,-0.1796981171418782,-0.9611107485955325,0.07356151613813054,0.3889237496665777,-0.5341145050725679,-0.3069175755317847,-0.2956980469221733,-0.5220326452991748,-0.43244347609387,-0.04424090575200106,-0.3426080856024073,-0.3052778597254517,-1.055604007795025,0.6269738487222393,0.3540179052783577,-0.6000344117021403,0.297178880509349,-0.5814765645574498,-0.5886533000709865,-0.5443194750866803,-0.9345849626262516,-0.2797382199858972,0.1428788142530579,0.07577974200351263,0.7950862292882936,0.8505911699351899,0.01159266178153895,-0.9944335079756786,-0.5067667980603068,-0.5453328251027594,-1.120545424602533,-0.6187649343197617,-0.1628236312537205,-0.3318053748897515,-0.3428436073426215,-0.1913939884336336,-0.1761397020184699,0.5516684925625359,-0.9481053851541534,-0.4184711900501834,-0.3872863154369743,-0.2916771406575658,0.02121797338655945,-0.8503568204870093,-0.1619956587328202,-0.02258394129773799,0.1540657030866742,-0.1584810427491823,-0.4893587477844418,1.637519980981871,-0.6453962155733067,0.08350998097994991,-0.4833795598090472,-0.01951667243391128,-0.6195372904548377,-0.03740274054497082,0.1239195525567606,-0.08138476805000122,-0.005652614899164752,-0.9121602698988032,0.06466548503518743,0.7834213569067833,-0.9084953766535383,0.6966796109064649,0.05933808483699022,0.1676932177907155,0.133634160154913,1.561412702662619,-0.4537992132349912,1.120374773262482,-0.2045155924178678,-0.4208439896151725,0.5913528694137945,0.051711254691248,-0.4355970455555015,-0.2225034754320398,-0.2105713914192944,0.1748976797685223,0.454544811507049,-0.04756713602037631,-0.6777863785843818,-0.3604666392991975,-0.714236767859216,-0.2324251716113807,-0.5766875994520702,-0.2405404132359916,-1.093398835549974,0.06147496739177907,-0.492967099502236,0.04256082012232655,-0.563059562873521,-0.3109535840067478,-0.1634733312433894,-1.28749686535661,-0.6661472272410658,-0.4291984997572773,-0.2265041782491908,-0.6613036312084232,0.6545545421988164,0.09173637341435284,-0.2481427829261454,0.1810071904052764,-0.3053080125203673,-0.01014291151143422,0.978446846050984,-0.562276901007402,-0.7002209925081603,-0.06743930407840037,-0.2093934209855292,0.1491113480939946,-0.4208283185976817,0.01557348171709076,0.09941583728973148,-0.03476349459669426,0.1470574507489203,-0.2934715916641452,-0.470751315470104,-0.2206367559439575,-0.4967631829642056,-0.05811778491304826,0.2550685562940826,-0.1891725200383763,-0.1062320496073268,-0.4313297581795971,0.4902840091846051,0.3685166919416801,-1.193638889697035,0.2682524814093747,0.1297849852589163,-0.217147072306701,-0.1826642666138931,0.08467241500882945,1.745210655084749,-0.389402631548955,-0.01939895051902693,-0.2135941930573803,0.07774779153964009,-0.2895369179348023,0.1180201361153014,1.123539490568286,-0.06227970179049497,0.813395995377122,0.4322438106492205,-0.3082462836765362,-0.1302944311495927,0.03161899395538657,1.414788392838463,-0.05138392978417594,-0.176430257356637,0.4286860969493166,1.286668149313002,1.686128326694362,-0.005564922107336737,-0.1984266142422769,0.4305394869933664,-0.1098233668058622,-1.303525801509307,-1.131894368170702,-0.6707064700649601,-0.2308195092267738,-0.2174748432253077,0.4973027423115064,-0.3830954550090724,0.2782743826046799,0.1418553961575673,-0.3219674871320083,0.3349835459349535,0.04544080050569154,-0.3298252857604746,-0.001742771808588531,-0.2545768545986394,-0.3680232560627233,-0.350105292055017,-0.451452797405537,0.5453326100867734,-0.3910121915712442,0.04858802973981973,1.100534799188067,-0.1034136163492789,0.005945177811736605,-1.065562132177611,-0.02363277545643457,-0.07085582610234281,-0.4174947553431058,-0.3972237122043331,-0.6935761318131425,-0.2981561538616788,-0.352615428627751,-0.6206004305550953,0.2219038755412435,0.06906566319280658,0.00417387441925663,-0.2455686421491441,-0.4396442957082291,-0.8364923648707039,-0.6576207784537891,1.110987819446746,-1.172794333518171,0.4431174848300839,0.596492393838044,0.2462732320226747,-0.4777092285521345,-0.8304443294344053,-0.7486786447441189,0.2544140648071996,-0.5057690980701726,0.1341367104520068,0.7733230625469165,1.229510265678256,-0.5419142267723892,-0.5225534743809204,-0.6470524124800808,-0.5709069490388207,1.029752892738704,-0.3044747501325589,0.09519949599070598,0.01877389620889676,-0.1236093641305697,-0.197746417301706,-0.01518077212677109,0.1753600199526689,-0.3274267626157045,1.446828117473986,-0.3472485663854581,0.1576506778702784,-0.3976639048118591,0.5144233332954837,-0.2357555275334405,-0.4645553566596118,0.07158432033897295,-0.3050655013045934,-0.04559484475033917,0.08066518471061916,0.3493964577644168,-0.7896457945627221,-0.3293589905855726,-0.5442113467978091,-0.00388157370953828,-0.05475861970017339,0.003769100253141773,0.3233898140895544,0.1589892758036197,0.2106187784541343,-0.1201775384631465,0.2053140943268335,0.4279566200631359,0.5225814124858271,-0.5335664302629547,0.2366115475552501,0.6300233809256141,0.3128542623149825,0.1493852985279641,0.2097302106603218,-0.06480582819004885,0.7630249062893051,-0.4976597054207628,-0.1105801050557385,-0.9509194875588263,0.2184262215606853,-0.2918583631821817,-0.3841429708184229,-0.04623817372103606,-0.8284527592289365,0.3790554354610756,0.2340582204296275,0.3064904015400382,0.2108052253969631,-0.6300054893172341,-0.87080571690369,-0.05836974963586018,-0.7227518449870879,-0.4917191827893,-0.1238406057598681,0.2780126460584694,-0.11024865463369,-0.008925373664481083,-0.3182827609057324,-0.631798203834524,-0.3494632101076832,-0.586708857162506,0.0422008167857111,0.7257412734659429,-0.4724713949141872,1.56522645281597,0.1286846555992661,-0.3011886878117915,-0.143629775689127,-0.07822297733055998,-0.08189088784737751,-0.2956493101292226,-0.2975881380740398,-0.1279685560101251,-0.3498536379560717,-0.5002202773566466,0.1735369296772218,-0.7916023865546498,-0.332060930803305,0.1240222679837553,0.4597079203895966,0.3196193832493934,-0.4276576788320735,0.8097579142005199,0.3637583389163994,-0.4529971359614977,-0.123438867631998,0.3159832454969733,0.5331455921177327,-0.3186879480309678,-0.4795241956186672,-0.5591861693332886,-0.6400378558204363,-0.3015547813554029,-0.3838843441996047,-0.31352130902052,0.4562071705982081,0.09198073187173665,-0.055992674838755,0.0483700308441086,-0.7108428191566781,-0.2333889426417024,0.009096567396323281,-0.5686210194337123,-0.6234646704350979,0.02545670006623587,-0.2187608101803904,-0.6983847046590794,0.05967281041590617,0.7514480870790031,-0.2721293689767366,1.384786030472592,-0.1122890759417132,-0.1059527758443266,-0.1942556730102601,0.6142609684533008,0.299548098224185,-0.2784375902784239,-0.0316799603449834,-0.3263702139279961,-0.3132228941283045,0.1046175040929259,-0.6429219274875623,0.4847197644604888,0.193575911207134,-0.6124716205859524,0.04609490261819673,0.9892105789856679,-0.6719226646667311,-0.1202534893449192,-0.4011663520128768,-0.1361484281260018,0.6992454037701354,-0.1275245365585502,-0.2718238619631038,1.111998409737158,0.4979751666291898,-0.6311574317767371,0.4269788744315912,0.3836321363023432,0.06460117670837204,-0.2624510935354189,0.1436946418064457,-0.3834394507387548,-0.08881524924663335,-0.3077809947753538,0.4979578020322539,-0.6154264051609517,-0.2873871079615991,-1.045815762645812,0.371038214888623,0.03251531799747173,-0.3311106426986763,0.04811330090984835,0.2066628398105188,-0.3850895244064401,-0.08482332585195908,-0.3908333104376497,2.18915347021744,0.4763463926403226,0.5537583449340756,-0.3304364608588769,-0.2114649368162689,0.5974539707741036,-0.2372621180773904,0.3411620522622913,0.06892362199072138,1.375762306091655,0.1980345308931428,-0.3252754549079592,-0.7665158889434254,-0.3848006367798359,-0.8083608246238732,0.04950789526204331,0.04049955633675436,0.3018493740349773,2.96381746540111,0.3426391302423939,-0.08961763826828879,0.2118711611271542,0.4575763607257229,-0.5042954825433511,-0.113276256869844,-0.4022038671253462,-0.08014517819888624,-0.1941408059734644,-0.5526874521525184,-0.7284306907310953,-0.6057829766936431,-0.2793195257846323,0.4191831075543235,-0.3124133359241876,0.2337184209591593,-0.2988659025264162,0.01724556872655085,0.04481056725595809,0.1991471853178407,0.293756945789843,-0.5156646984082566,-0.2004149233473174,0.7254735723641565,-0.01044988583656214,-0.103687766731204,-0.6352691474742923,-0.0716900163296121,-0.1433870975043844,0.4347789691696806,0.2530773107148666,-0.06951225177740303,-0.146944415126526,0.08587390223082478,-0.4827303817721729,0.5129660212986996,0.4249930404893347,0.1237976218290766,-0.2717857693792248,-0.2062573194999616,-0.08615672572991108,-0.2792967745404409,-0.1041942105023717,0.04017523072661113,-0.08116343926903463,-0.1329868428252685,-0.1211673130125248,-1.148368226514804,-1.428955882798765,0.5703162323404189,-0.8196731389334734,-0.6622652693460119,1.297235857880305,-0.5205974456510168,-1.03343165588639,-0.1590834195552469,-0.05957913828778067,-0.2991228100493283,-0.3402604259949618,-0.2770461533723785,1.755051953643063,0.03329623200464332,-0.06297766887815734,-0.4792559704348084,1.313551577867059,0.05676100061807597,1.128017450181203,0.06706455589249002,1.645313841762029,-0.4330682902927105,0.2887146886204328,0.1751210981670306,-0.07437126017019656,-0.3159193402975219,-0.2527818843771414,0.0876950325761202,-0.03141085255121177,-0.05487475935418115,-0.1448681791129175,0.1296342371537838,-0.8200733782379801,-0.07546008871247843,0.03943558566474231,-0.05037692861502057,-0.6943200081360754,-0.08403581534607824,0.2622517540505152,-0.404678138503593,-0.1666682317699307,-0.2271590351914667,-0.04697954345815909,0.1313910766836409,0.06375606679647706,-0.5158095526736736,-1.523223652987831,0.2779733458740429,-0.05849552846118849,0.3977817284295203,0.2841103669814096,0.1447831161489707,-0.7200744631142053,-0.4266551335982053,-0.3781942726876226,-1.296902482899728,-0.8981054832325188,0.1062820467459321,-0.4897183162210476,-0.5781148001523436,-0.1895826393305252,0.956029001637658,-0.3313649587430841,-1.856238263261327,-0.1041351344659286,-0.2643963217778891,-0.139920178145971,-1.161250831040753,-1.192679953895901,-0.7461392331950663,-0.5583073669674418,-0.6529820444788482,-0.4677814967325667,0.1729087189149972,-1.729041911360215,-0.8384371985339734,-0.3011546641278549,0.4621078927442214,0.008504828887571715,-0.4641551522272512,-0.2531499427904499,0.3855438806328688,0.1338932289949816,0.02903012203119102,-0.3576668657627654,-0.436350832626139,-0.01937835552033762,-0.2154505659848671,0.3142721637424068,-0.224700566806462,0.1161797862155261,-0.702643185720891,-0.231783856930433,-0.1922315100920635,0.4068980417841613,0.281315185911201,-0.2301563279416498,0.3504526814061618,0.4649790141452071,1.745759197031213,-0.7436453409231993,0.1055935714225871,-0.4130337775745667,-0.6165254195139446,-1.183349311546023,-0.139758420422314,0.8718656684601104,-0.1462450077341739,-0.02612502472244687,-0.3324149336488187,-0.1210643485634477,-0.4147499137924634,-0.5608402583362613,-0.4299758456794853,0.0005508484471061096,-0.6680332055678483,1.314699213744973,-0.4075301204092469,-0.7260646516255671,0.6790132116700325,-0.289422186497398,-0.1939279201573747,-0.2865413291854331,-0.4447810777463745,-0.2778771766576321,0.4931875912863218,-0.4628513956602326,0.08196843714121559,0.7575862991474741,-0.1097076868271457,0.7018961197403503,0.09101133040747331,-0.4593860486696349,-1.021706212626091,-0.897945071312958,0.2283785832040754,-0.9099933567150191,0.08855391351607829,-0.1052148658659235,-0.3064207340448512,-0.4914258848892982,-0.69566656775576,-0.09754997347347694,0.903093778612538,-0.9718066813697519,-0.3976925175968011,-0.4848727075195327,1.463398128538332,-0.3249782527426362,0.1064074340154625,0.006574752509617737,-0.4849508440939325,0.1745030595927157,-0.2148434840603366,0.6133097972647128,0.3295659833257053,-0.4043133210674299,-0.104905271582051,0.09919495744932559,-0.03277500739485942,-0.9534319617319621,0.06392629573925969,0.1810110473912563,-0.01806002247782453,-0.1158530148698013,-0.02902706935376903,0.6760380307570962,0.4472650353620893,-0.255695067515711,-0.6437884812649095,-0.1767277809758082,-0.6739050494934796,4.917303640160482,0.2180277856457541,0.4511037110546164,5.515186339035886,0.2417864044933416,-0.2710639587743803,0.2658692804014279,-0.2878544139727415,1.173387725017299,-0.7265364916310728,-0.3366845109156153,0.1001975198008531,-0.130195149994702,-1.412078923882021,-0.7237895075106417,0.1972100841227102,0.2231335082005882,-0.6738284074633325,-0.330354736524993,0.1109813828809079,-0.3785500876962763,-0.01389535934158996,-0.7622666179546446,-0.5833689079824649,-1.194220885797356,0.7874022374730698,0.02710291479183513,0.4312188793700211,0.1131838382793001,0.0590657984039339,-0.7325978233139989,0.3912249414711854,0.9487693752499009,-0.3560017165679836,1.382698733208868,0.4897420446197882,0.1134357822313335,0.08315066252451681,0.4760379938174457,0.823527599807053,0.4584980117039136,1.099761925778237,1.496208537284488,-0.6393416669487121,-0.3536101531015188,-0.5316340986843908,1.309305075174247,-0.43618664908371,0.4041906772918742,-0.5717024871200195,-0.177858766309427,1.05968955270326,-0.1608370053059799,-1.005382066293196,0.1301499334800559,-0.2253609913505692,-0.9352147200664728,0.371295434961419,-0.1595956288797969,-0.0164693431024669,-0.1046539050813995,-0.3767152694151651,-0.6522945701045912,0.209449459784332,0.2733843901884579,0.1152503687296958,0.7798773076171834,-0.04830028998022109,-0.6964061421433424,0.4852504859574928,-0.3849313711362136,-0.3895273595118645,-0.1180885098698706,-0.4610007724629152,-0.1270625357608628,-0.03175651099930825,-0.3756513488211325,-0.09531107689613927,0.1194221190099437,-0.3053815618930629,-0.3018583477745097,-0.2805204515880649,-0.5662413561589669,-0.4183130639703105,0.3914466054810395,-0.04278966477631455,-0.6465304154228959,-0.2618247627731877,-0.01084171827473997,-0.4039702995682561,0.8762221096307605,-0.9059749472823103,-0.52528645660416,-0.02253243953429955,-0.303989997071465,-0.5761835623307787,0.163455903415124,-0.5027725875055096,-0.09940828820402385,1.095645083145248,-0.3565433486026995,0.454913655560969,0.1778965561282842,-0.4128154656763468,0.162355193296115,-0.3234031924244796,-0.1882646768794696,-0.1363569823393865,-0.3592247133449438,-0.5016956513932678,0.4302637739288988,0.02584677245486316,-0.4104707309311249,-0.1445318902528999,-0.5088395582486315,0.005626353613627233,-0.1871050564336817,-0.08365389273817948,0.03237362417806634,-0.1426500193633922,-0.5573085287602795,-1.088806688924748,-0.3489847099293704,0.2628055854391303,-0.1307893127733574,-0.1177625005976336,0.2373446443125451,-0.7327093578058828,-0.6205614959180844,0.2232852927222788,-0.4388715173115753,-0.2437933656801638,0.05701110488989702,0.7192447608637966,-0.941124279099312,0.01352865061489728,-0.2033756986666954,-0.05290525242669224,-0.4738612459432572,-0.4333490528820829,-0.1213606078738585,1.029854667276204,0.05659935263248683,0.05410066630111783,-0.5561803262062919,-0.5557986413888797,-0.1546180395795823,0.242251608856409,0.5027477309670526,0.1690794583056034,-0.5858480765656346,-0.1982473571274742,-0.6976474255111889,-0.2319531826055571,-1.023302146328306,-0.2740789011078649,-0.03479696901939369,-0.3343890661943548,-0.1478908816542008,0.09532161095870098,-0.8924103466597267,-0.2543390287956198,-0.2800206274104538,-0.972155390879342,-0.4270376667851966,-0.2568933625227501,-0.1069867985065458,-0.09573308625516193,-0.6024866959275077,0.1332804009954962,0.06079496192993331,-0.1064289518321511,-0.1731249021023287,-0.02968049664859375,-0.18948631500338,-1.033907246130626,-0.4650661213187056,-0.1292363455258904,0.1709227008354588,-0.4616275435611565,0.05895769937825372,-0.07026071917164294,1.329697325025073,0.2876470679887531,-0.01866231846261337,1.927180122362554,0.08284346804023705,0.2713447324638186,-0.3345971965851308,0.0114986558980015,0.07346082641524024,-0.2771664140229095,0.3788456925929697,-0.5665498583337542,-0.6144329762759356,-0.3160864551111488,0.1064769540011993,-0.2163682969280785,-0.5157637084353669,0.6083656302786651,0.4050915961933959,-0.005156001625017025,-1.035991912868361,-0.1308071942380498,-0.2998841329470681,0.1987868272150269,0.5998785210265539,-1.588124401566483,-0.5428088188534574,-0.8441793401470744,-0.02822485362481606,-1.795058204894632,-0.1822780796384886,-0.1473658349271303,0.3398241196433159,-0.2884032391943074,-0.01521477738118265,1.803780311336257,-0.4524528704448704,-0.05090714158988954,-0.9268451742880494,-0.008943195213621415,1.599220873220063,-0.509496971061937,-0.129089866583065,-0.03670209908968725,-0.01569011733224867,-1.288697051817174,0.1932044176936442,0.3655458210154794,-0.3646836852318834,-0.4747766818052992,0.01363612780406332,-0.4085866182649253,0.1409281012425593,-0.4390240862959967,0.06509323011314316,-0.4256527675712756,0.0004013552122903219,-0.5938978584713611,0.08672417992253333,-0.1451812104235335,-0.2158329141543784,0.2287645487646571,0.00376236436648483,-0.2023632069249773,-1.166254618697028,-0.4978842100799965,-0.2848813201988277,0.6399955103941778,-0.04301507380508175,0.2068032154000224,-0.2265942013913586,0.07486629023964746,0.2675822586785179,-0.3087235725883933,0.4195557945587652,-0.5974837384065537,-0.2600113007938422,-0.2174095444780972,-0.3655525995794879,-0.6679062177866898,-0.05433091338508016,-0.366522515216495,-0.1277753400300752,-0.1373738689158679,-0.5951983055901097,-0.3157195288194777,-0.2603092817018675,0.8564265538616463,-0.05283194793964268,-0.3569200472529053,-0.3560756048508794,0.2540355700540866,-0.1286628193001733,-0.1826794459889854,-0.1713425604350877,-0.1483645664343562,-0.08117442621708004,-0.7361786884588214,-0.7873852743443855,0.712784605621817,-0.8706123856330303,-0.3631662565613576,-1.489044981532286,0.637466455246488,-1.148207800476038,-0.34994603989273,-0.09095821827438635,-1.071395847089117,0.6383682831385283,-0.1683728665697165,0.9168343035462923,0.7504062057167189,-0.07534483130127724,-0.08046947058485669,0.3031005780877573,0.2363324717920624,-0.3456516526927234,-0.545615975287394,-0.3903797081466713,-0.5507727916638505,-0.463306815519706,-0.2746273592307004,2.135009837322673,-0.6615300146026418,-0.4775780558289593,0.310420961390951,-0.4398948298038445,-0.5006490897698911,-0.02429950909397305,-0.01624945178273257,-0.1119153784692209,-0.2796707443877028,-0.06547514469585308,-0.4157894166437148,-0.7391043065929795,0.4136888986592071,0.03779674405273705,0.009304205536088263,-0.01789653095065537,-0.1129983267779169,0.914155770308354,0.9936984980296043,-0.3985869104565041,0.7518302119757642,-0.3600160772978739,0.4861239849254525,-0.5268272426117924,-0.2728007257091134,-0.27194693713398,0.6408776216582409,-0.4508512020181276,-0.9797396288780083,-0.6082072169631088,0.2322237784729484,0.01257576526454521,-0.3159896872398955,0.6357716026660578,-0.4661072803214374,-0.6054911263336098,-0.8186720752756349,0.1288201840933633,0.1380559364957243,0.3108656463871059,-0.3016079766105075,0.7372780055384013,-0.1551992883816527,0.2532372779983305,-0.1253232400258771,0.8206893638404203,-0.097531622117045,-0.469839863836625,-0.3833608803580096,-0.05147860790200167,-0.9037524880027092,-0.0253921508486281,0.6978926847758213,-0.5953020647824252,-0.06945182948984858,0.1262975824633489,-0.5550298884012411,-0.181453313237692,-0.6181781254591254,-0.2438177594093265,-0.6399112363956537,-0.5623306593038291,-0.2362246302975015,-0.1988800908074841,0.02739049017083713,-0.1256701449143773,0.01154153280767564,-0.2266139966890577,0.2109940239337728,0.1655336167622038,0.4041730194565175,1.51116338829514,-0.1625401582507885,-0.1320566814020149,-0.05009422736974071,-0.1520919994393256,-0.1045857349240448,-1.349082384706462,0.9990529487948863,-0.2059858773754043,-1.841040150100351,-1.699263760588253,-0.2586991107632775,-0.1892363835897123,-0.1971207388482842,-0.6269020326926321,-0.7012340333081153,0.2684018179325822,-0.5452051217563171,-0.1579775570314073,0.1787147787359411,0.2458080094513878,-0.03410493910321983,-0.0431710952137323,-0.2644073336715188,1.212786661624861,-0.1525870739000729,-0.5867629127471055,0.04084810590717106,0.05523476752782148,-0.3846937522811151,-0.008347897116794177,-0.7795135940738014,0.6135585793465347,-0.5013478585030372,0.3984986476089427,0.060870081536456,-0.559143267134585,-0.3181482715065571,0.339175475323395,-0.6143407794879989,0.1124342575674035,-0.7955817364544494,0.1606422617414877,-0.2578827037382097,0.5819977851338659,-0.003512779585515444,-0.1371579136396363,0.2271895890791495,-0.2742582340283679,0.03458210998994658,-0.8789343279832276,-0.04615867807281734,-0.7656000782813847,0.06548419697217754,-0.2142732782823139,0.329003104136822,-1.043345364075639,-0.2489399295859591,-0.2251916935604532,-0.2868680283648005,-0.4026821418415304,0.04213572235468165,1.424437687932839,-1.2936269311374,0.2421698483725464,-0.1130079762044113,-0.4375547542845901,2.092151557841797,0.05427676744061839,0.1986113638704666,-0.4326033129587075,-0.3135085014162021,-0.1311773981595895,0.1583642313072433,0.6820317855355117,0.4273168132204741,-0.5603036175990724,-0.2857858675893352,-0.3594972843884865,-0.256498984256744,-0.2066335679430856,-0.1381668158495992,-0.07877429299589955,-0.3988917848049198,0.5897787662311557,-0.3547251160403244,-0.7067891870280103,-0.2060681997639878,-0.0136313283777415,0.1194920096973186,-0.4830527486435612,0.3969715364154947,0.1158721834754062,0.64096221203387,0.02612579494248867,0.3755174301538785,-0.1951027460670177,0.2062023971207568,-0.4364597788460893,-0.3008362466875709,-0.3978765909589076,0.01487625138781773,0.007403585329145978,0.2291044319253348,-0.1293850622495457,-0.3299296714015854,0.3240896875393296,-0.5028592706903444,-0.2106451549677554,-0.00145749627597878,1.801381996630483,0.4531269033023552,-0.7603836839256821,0.00807574490464332,0.06519599397060617,-0.552785596741448,-0.6158489935656348,-0.3545010740573276,0.08923612397383313,0.2284753851287936,2.0784161316419,-0.1149589094053299,-0.3959219932629884,0.09820874224782493,1.320412028814379,0.5389785076551541,0.1028980802678139,0.9516442273064687,-0.1029854926987766,0.7527009591282228,-0.8568792968137042,-0.2185259506319576,0.0825282355666018,-0.5476749548865159,0.01567463366154366,-0.1850327321296673,-0.2619513495025095,-0.4190305504640047,-0.2491238032960395,-0.9107048760589563,-0.6191069579115323,1.020617401164919,-0.2178326848051428,0.8130597356025084,0.2095661010108636,0.4016081628694634,0.108515415292823,-0.4400313425123289,-0.4038513735357314,0.4248543899499083,0.08214725888839565,-0.3381220713485181,0.1810548842287759,0.1201540876514594,-1.053774367384615,-0.09830909574948656,-0.2188326774011313,-0.7034439581926173,-0.7176836674875106,1.267776185833357,-0.6561740341755578,0.6505984725932532,0.5474966514732481,-0.03522347845398341,0.2868689515545219,0.3552230312683417,-0.7412945420418835,-0.8077480514385552,-0.7973508625366913,0.6939750627871951,-0.3098961528644678,-1.649501316218018,0.03300889292863409,-0.9952693742547226,-0.1368667159230472,0.5061848119452228,-0.4840023857202818,-0.5048886663937177,-0.03291304713449057,0.5014813304164284,0.4604922577543344,-0.01350752229062786,-0.3675402990495951,-0.02400631493713894,-0.04402943718576264,0.1731529144170586,0.1520960524744274,0.9614871866705987,0.6762169029102784,-0.5136660268121404,1.026449182147517,0.02463230624861962,0.7964341085760075,-0.4276951713279445,-0.673875467948681,-0.1890602101881858,-0.1124119781981231,0.817884494892776,-0.5894000166432558,0.2112213932185242,-0.603235237516013,0.5243713803947204,-0.5902903578797205,-0.5404204562329036,-0.4636656922319531,1.385619420774475,0.2090321349351953,0.2645383780743769,0.8718147277739974,0.4888725292612389,-0.02556895654344518,-0.2406701169048721,-0.2278233015039964,0.6593214798096569,-0.003718025051774161,-0.279564172939385,-0.3586331845677608,0.5891081244217325,-0.2279920971220585,-0.4249766262810815,-0.5826172423844475,0.05262278709366344,0.497338298488296,-0.07752800909747108,-0.5912869062080289,-0.2279036952881761,0.2707980687783742,0.463183513206479,-0.2292072417885783,-0.7182626776436321,0.2910501209429115,-0.1880259898387381,-1.177520758520464,-0.2277153643986541,0.08554564847322897,-0.4089014925845438,-0.203213283059583,-0.09720435851646415,-0.2535063279459561,-1.478872756316771,-0.1227217691266637,0.1326834898397328,0.1120911389941621,-0.7415559972715333,-0.9429368225070335,0.2566663926071943,0.3032022776591735,0.1071504449276212,0.8248700194646368,-0.205559916597849,-0.02374511287895294,-0.1792711212149094,0.06266983845505031,-1.417412907111986,0.2416982695120329,-1.276996815540642,-0.6602110397437994,-0.7398006718157326,-0.1263116434900937,-0.5310875897910394,0.3070715902732785,0.8329873148004339,0.1724173726399547,-0.7235474164240201,-0.0499878597591992,-0.1786387128402533,-1.225784634799506,0.5599705180902925,-0.2383965284795921,1.027789419677437,-0.1647256537384857,-0.348095121583659,0.143794290585479,-0.4193782475016516,-0.4477795275672316,-0.6144960023191008,-0.04013438789072638,-0.03642014221390506,-0.1725809575834268,-0.5935557840617071,-0.2930662248792997,-0.07770094183819468,0.004398615879470157,2.077853071057538,0.2854410269611358,0.2845382463806569,-0.3661674761823928,-0.7319843512034721,0.4373517460758389,0.08727936473823916,-0.499700351811869,-0.7943730650303938,0.83682863289429,-0.479920570500132,-0.2326133735022229,-0.326110548123276,-1.078367352513623,-0.5162440754246002,0.7406279655352995,-0.4004439075859534,-0.7427157544171238,-0.898240507596904,0.001481112217309685,-0.4958582160234292,-0.3238167963314527,0.07265829967827302,0.3916894848066535,0.2165288230169949,-1.060081466706398,0.7531468093782622,0.5422518055198371,0.6704681896324028,0.8147181512457057,1.179350218958622,1.054211424708708,-1.318505732727413,0.07106989585638822,1.006486383953641,-0.323982926899498,-0.2643257348541842,-1.679848187601149,-0.490031600159501,-0.5270355193573442,-0.3111194162523567,1.292003161875736,-0.1894507252823609,0.4416059655269027,-0.07723620933234664,-0.6202081279843336,0.1847218792958437,0.5429920013141023,-0.7802207372382488,-0.3701119065614308,-0.5762724319609447,-0.4679802403974718,-0.09221424341838243,0.3242815618600818,-0.6339344653157002,0.7209550750808504,-0.6152168804883267,0.3816227334456865,-0.2136717368862611,-0.4780075776543746,0.1731022481626517,-0.427455227947869,-0.1781193939128879,-0.1197503617412043,0.532765961944751,-0.2164234123894235,-0.05677398194252795,0.04534407683028023,-0.7629681975562425,0.9791096382279662,-0.5215241564736003,0.04712785266889302,-0.688232036475331,-0.5280862513829306,-1.256921810942293,-0.3999581991611039,-0.08880621566401818,-0.1057193161112539,-0.2597288550887047,-0.7809170157590696,0.420226921592777,0.2308254332389145,-0.8320102810545221,-0.2532964084947821,0.4728103275829668,-0.1753587022022012,-0.1245226099311788,-0.09131017104326127,-0.1585483599200496,-0.0817386198100569,1.373861832535972,-0.902788365673561,0.8647114928588687,-0.7640909530343082,-0.3920197437955475,-0.6368669576183724,0.242572573214952,0.4096404385094901,1.842884597620127,0.6940818312859377,-0.1044336795140992,-0.2325769424294235,-0.1923163844174965,0.2954289641187741,0.5247065857336451,1.869632818590597,0.04098474646721104,0.06939472143200405,-0.9545488923333276,0.4527860931077101,-0.422916096002777,-0.4049993331848966,-0.6173237377956309,-0.2207197304283785,-0.291918155990364,-0.09783932943822843,0.08844073550081206,-0.2371162706889454,-0.3017712986164618,-0.3385143697887571,-0.3974945857894832,-0.1322251359334072,0.09811765298032414,0.126480953125627,-0.2410296616736268,-0.5292872114247071,-0.05742498249117967,-1.388464582876153,0.04000611820173307,-0.27119195707052,0.2234285096219317,-0.9837845710980117,-0.3713560981722562,0.4233948601429776,-0.1773299956811328,0.04530473226785935,-0.3140174116160988,0.4543745388210799,0.06955233341704568,0.184819558430878,-0.2851482041850295,-0.5357646796188624,-0.0608450473416306,-0.1558441330977086,0.1258767845690198,0.1745855724509363,0.5245207550468053,0.2846284984870279,-0.7893749586807369,-0.6702652084541334,-0.3575290140383217,-0.2884390649612518,-1.544555402714184,-0.1299637258623706,-0.07222966179556942,-0.6913471862627804,0.1551337293713144,0.1718306072816071,-0.3731264433890792,-0.7373174498508654,0.4760811937790463,-0.287102499181759,-0.2371940572414587,-0.1210707581585757,0.2646352573166544,0.2131369005079021,-0.09490914149824502,-0.4401310625057954,-0.4586770307430217,-0.6894363912461242,-0.7534857297734573,-0.05539696879424932,-0.4913011341421951,-1.230770703677165,-0.1658030930484645,-0.04113760863503174,0.05618196327629889,0.312377348419042,-0.2187790520569081,-0.2445075275027859,-0.4290982534014847,-0.5209340681685053,-0.233932306421772,-0.119424360269976,-0.612951142496026,-0.2718172108135004,-0.002485162305151973,0.03735537251015213,-0.5727354351963786,0.759617594483985,-0.276578639100942,-0.007494526721473157,-0.4041229723085573,0.06897954238285564,-0.0003507766453837042,0.5950711684962869,-0.5408243141422874,0.394775177432242,-0.04537520035704759,-0.1736308445602033,0.3183941139168646,-0.7738909544012482,2.204392149416734,0.00946291853228174,-0.09152147063489097,0.8301382053541859,-0.5029507199390028,-0.4441825210162184,-0.3082166277988465,-0.04264343087287373,-0.6267270805765845,-0.2334058633364156,-0.455598097187916,-0.07288815117553486,0.2215210796471053,0.4276746440704029,0.03631462078794336,-0.7415574858945007,-0.4108562458955782,0.1971132558223802,-0.4174485265454324,0.08859060271312764,-0.4877912610623946,-0.7198885260224444,-0.2501887908706744,-0.1520389886278892,-0.1812596274676792,0.1207265457720878,0.2585771205516616,-0.6119946834459423,-0.2753035177041471,-0.263422638507835,1.824990753543472,-0.3780411435563349,-0.1001843584874782,0.6421989232609557,-0.8087051194672742,-0.03106777237212175,-0.01327246243957876,-1.26369877611303,0.5785666218238487,-0.1065644365516906,0.1105493518951348,-1.666749274918534,0.01963337832644908,-0.4063381003988547,0.1173851596288775,-0.3920501925839678,-0.4760459728715142,0.02123999439165006,1.977924498869205,-0.474676566162631,1.679063806323197,-0.8815161417115402,-0.1125415369964574,0.02291316335624416,-0.2923991384742926,-0.1252771704475274,-0.2180766437149141,0.281123467287091,-0.3191068670098688,0.6732873075816134,0.386055602291153,0.2130465104860859,-0.06038983232789631,-0.2828239769310771,0.6090541314966254,-0.5608660744513045,0.3657230021589901,0.2330526871586354,-0.3210220092101262,0.06607805058609174,0.203089892998698,0.5809223821117862,-0.1049317424629267,-0.3254232988001333,0.6395355974852683,-0.6964446009755881,-0.09941745973625676,-0.8452593107815649,-0.2714865188411179,-0.3682589626317759,-0.08315484057244776,0.2517572532446278,-0.4751564852840895,0.1638742053340199,0.3864591940250142,-0.1588871620693524,-0.4102867864449267,0.0005861203668831492,0.3671993607995719,-0.0531468857070589,0.07554865449448947,0.6280616288770878,-0.3030358914813892,-1.011827869345032,-0.1907708057997081,-0.6802587153517397,-0.2476875177929494,-0.4233423962537844,-0.1210707558106639,0.2781434521583496,0.2291862288704563,0.4303160227123253,-0.7664789398834152,-0.3096039438145702,-0.08989565191712005,-0.2955676576454324,0.7631250028987957,-0.2476655221052557,-0.3452764902832262,0.2879739919046285,0.5270965564911638,0.5792553326471294,-0.2665454056809712,-0.07698337500890838,-0.4135150934611211,0.4970826949365234,-0.4518246028645256,-0.240855463975243,-0.9054326834172973,-0.8007671276306711,-0.167390792255679,0.7330168392199671,0.3390256523969054,-0.1842752806879986,0.8032311279249661,-0.07212412094247588,0.5968904101535502,0.1608837669268163,-0.3256792449363505,0.616993437341233,0.5634280589078494,-0.4403728248720328,-0.3987917718332534,-0.4587105508261241,-0.7132488920760347,0.7225061063212361,0.5919961682390262,-0.1152691651205639,-0.2216876085376398,0.1945937422386351,-0.298322227752739,-0.325108163171891,-0.2458589582792683,-0.02353590908220707,0.7217169439169547,0.4774257303970994,0.5503723888566834,0.3831597716045573,-0.2331770219058948,0.6567836741866137,-0.3703946640924762,-0.8364111375749488,-0.444098899021546,-1.288738005443142,-0.9862004424173882,-0.5148486526671497,-0.08366109087019677,-0.2011122042897213,-0.9403423061558999,-0.06688432355695884,-0.3148097699976838,-0.2044184465511653,-0.06630689534148619,0.08574987447525323,-0.86284673844131,0.1759735460516548,0.115135511022003,-0.01226892407550144,-0.2155416664975427,-0.4677295636623211,0.9430254161414182,-0.3254774744868907,-0.2913473197368524,-0.5064624160727332,-0.05794956568670128,0.07093626447010458,-0.4515782761201042,-0.2142226346546813,-0.1934352065191842,-0.04830812649456895,-0.3422521803085089,-1.02784438493344,-0.3374307336230248,-0.08709393650497609,-0.09942670095178946,0.5760819215419647,-0.05744282936111873,-0.06986210313279044,0.1454596190478585,0.4071154810261771,-0.8306975269741231,-1.216779002628171,1.026521612804824,-0.3211589568019369,-0.664338074466474,-0.0341198518690853,1.480474833320496,0.07573487112882871,0.3360405527985105,-0.004218144805302149,-0.282886408440297,-0.1609478821209105,-0.4698513167768502,-0.2401696161372234,-0.009566999784624492,-0.03009376251035402,-0.1434542171076266,-0.4729920406123419,-0.8297027544665627,0.889682782192005,-0.5495415468146011,0.1258867974620048,0.09049968894247709,0.1222807521072564,-1.048733951990506,-0.4902220543732302,0.02142992390065231,-0.06371155778093988,-0.6169284326826615,-0.1407606731407753,-0.2723656980080899,0.03671895799066897,-0.548643580895487,-0.9015597828193558,-0.2790567933348351,-0.05940216892204209,0.1552385467620692,-1.790925929909389,0.1193075489275346,0.08984088771423843,0.446884079602564,0.1999532560530744,-0.4716166845060686,-0.6241260804641522,-1.052080422257358,-0.2275277602387333,-0.1844155286974277,-0.02072306446525213,-0.5042919626862263,-0.2588848210014049,0.471050891539551,-0.03739611167875272,0.0001753374805635906,0.6387417767652085,-0.1071388832543363,0.2267480041514154,0.2278461627502115,-0.6794236117661215,-0.3965210470845713,-0.3623200545376618,-0.7942902621779985,0.4020629691805215,0.04965146650905289,-0.4138630180163976,-0.8783243249299937,-0.1465180493404494,-1.077290591081155,0.3724883509367788,-0.363604371697787,0.08902158006149999,-0.504309974480678,-0.4551639006638067,-0.0232013393596738,-0.0002150738543130058,-1.112472612921723,-0.6268470280724153,-0.1186518232347803,-0.3694397142290247,0.1705365647722853,-0.4531516035644566,-0.3020967526751119,-0.3347745711623514,-1.731346183256309,0.0808362509843456,0.05663977209404134,0.251322246538062,-0.0624828932163719,-0.2998404234948797,-0.1490992377749626,0.4315899654797721,-0.0523122683174146,0.5325098988576846,-0.1101218922805731,0.4986510835709471,0.8636251413035745,-0.4173313721960636,-0.01080847781637437,-0.5711986437002463,0.5146078391995184,-0.4434321215228645,0.2067308488495119,0.01617988474520027,0.04726723720543856,0.03564249052842269,-0.07901370391814842,-0.1013302488199549,-0.1612733254415431,-1.048678606427884,0.07590223242124937,0.5204280468110517,-0.2419753703426987,-0.09407144171991152,0.3437590039790323,0.3581575830960059,0.01849485416449598,-1.149763282678545,0.005541500711096179,-0.7491099908043305,0.2178970749525483,-0.6865921347601806,0.3427651622615899,0.02390663473688577,-0.3579276250113863,-0.1171601454931277,-0.5621903636654459,-0.4234732724333288,0.02982956112261499,0.2489867059923943,-0.7273764734177391,-0.7786726451759219,0.6464412163070415,-0.303677503062376,0.5878884302036966,-0.2261135284357331,0.2514688122329315,1.300580454880732,-0.4483416565421175,0.4079738779673083,0.02141185088834285,-0.3811254174846314,-0.9305020286104655,-0.5086945726367411,0.06143281834005011,0.3671765134897294,-0.2772809382531201,-0.4182908384993725,-0.23172008391153,-0.2727540579901094,0.1286306888336597,-0.4323758559044729,0.6004616828355062,0.005644067425500944,2.064320640950459,-0.1854377822038282,-0.03781053698670409,1.74692015007282,0.3329562149038152,0.1266559203795974,0.09467657999651793,-0.02550632483017063,0.6325831265609831,0.08218527574462099,-0.03599723006835512,-0.2533709736933465,0.7967641212873074,0.05796014411038043,-0.7068778898885834,-1.606081217162206,2.504755581766852,-0.2533723487227899,-0.03263632094841833,-0.1056451088680423,-0.2480428673334057,0.1700616864124427,0.1493296736334858,-1.28991092556882,-0.2911435927339266,0.05070607554535855,0.290531246815977,-0.5862028072789748,-0.1039817522623084,-0.1386533362124879,-0.3405563474323033,1.184342007227714,-1.295789325581276,-0.6443940088359956,0.4535958602317219,-0.1770843016191747,-0.37690162225602,0.08412239356522686,0.09378472040530331,-0.1179593734452684,0.5961572921294251,-0.3562733056474257,-0.126909459544202,-1.196517733740655,-0.3054746999753833,1.295317919988552,0.1805945588345539,-0.02834089744236797,0.1682829619520183,-0.2719203143416543,0.3597673948887576,-0.2902387878996013,0.5184796511070087,0.1969754792492308,0.2400782610932609,1.147795824106133,-0.5653020845740504,-0.1475290549922706,-0.3671298620193513,0.1154860040743347,-0.1734714498507358,0.02136075630928989,-0.5540696224519354,-0.08705107026236424,-0.5763736196583819,1.318895192913558,-0.8383125995345553,0.05304399793500763,1.191079070768883,-0.4101985286139683,0.1862792321477096,-0.6481206821789275,-0.2130134072500507,0.293732220531719,-0.1948063259105682,0.1602508833939638,-0.5378989107497939,0.01430737885032596,0.09805350546789289,-0.2622026652070303,-0.2358164520395044,-0.8907928882097619,-0.1688854935824106,0.5433388684492352,-0.4606696355824956,0.2327810236340236,-0.3007723197431231,-0.3118943542326151,-0.3411840002585015,-0.2605781933461179,0.3190989231289142,0.329642878742924,-0.1762581545291663,-0.2202684524030532,-0.09106568521641478,-0.1791502519564069,0.1908747138205574,0.18884572615761,-0.8691446137047819,-1.335204650680512,-0.1671136894095245,-0.07128439732579329,-0.5616826166471566,-0.3026191894697611,0.16771472049944,-0.4258333796223553,-0.0855694298679386,-1.07912105396387,-0.1165057303719071,-0.07809830365079876,-0.5096289150838517,-0.1232849910175061,0.01511438856342914,0.1379250283350792,0.02491969604066308,-0.2618025841568689,0.4124162270099391,-1.13697878653372,0.1229229818369044,-0.6377457174199749,0.1659497438885085,0.1428959929120308,0.06571211313427595,-0.1257447780517549,-0.009771891524233651,-0.2673706652363786,-0.5019152209122275,-0.2756869685683351,0.6187421894377146,-0.7337598655584069,-0.1297848721328355,-0.2744174099527854,-0.6690996017264478,-0.5831688521203156,-0.2293327293555348,-0.6561647039067742,-0.02491701342748382,-0.2468144119331537,-0.5522697730988841,0.1720432748984199,-0.288368604959936,0.08644522386453758,-0.4075979066828813,0.2059492235168618,-0.08832717595632428,0.5411286195266595,0.3343286108028577,0.001839167491358339,0.1131339362076379,-0.6427302308267777,0.6285816222396761,-0.3837883968529428,-0.7018358926395262,0.3440147396724142,-0.4361094340600132,-0.5103053253513121,-0.6604706584084378,-1.178682243312388,-0.6687879471194415,-0.4076323449671727,-1.068313349012452,-0.260508759890469,-0.3656008017756466,1.322660147769967,-0.2226282618377904,-1.4744204356269,0.7997337005234425,-0.233171853886732,-0.1444825289902866,0.3724893542748067,0.1764079833749154,-0.1624168444062022,-1.310530923682384,-0.6065728875904987,1.044315999230693,-0.1291069898904376,-0.07024430404887867,-0.1364147282720929,0.267753465912526,-0.113120773321173,0.01820320580798128,0.8356627111224814,-0.1793568649708892,0.2549382803366481,-0.1461351429030201,-0.2352858819382463,-0.1753053382884597,0.534479979142948,-0.07754395093085442,-0.1340990990342265,0.3973250150963865,0.07712721172077718,-0.7442249941791644,-0.4216748258177124,-0.1384858760688,0.2971344959581188,-0.7806745188483764,-0.1608523233242872,-0.6776006174449622,0.9146477653289267,-0.1992268487121828,-0.07578467111792384,0.101140550098121,-0.306549888596367,-0.07996714750884006,-0.4081902766040683,-0.3186327193821833,-0.03363660060331242,-0.5293773301677541,0.5110210853528525,-0.8352659490901654,-0.004783917940126959,-0.2540892549151332,0.07754965877025587,0.1079501850404676,-0.5767396213291235,-1.229020401596089,-0.2641622413760246,-0.1144760607597499,-0.09446186258288668,-0.7243256447080145,-0.2855316402940826,-0.3118045776015279,0.03030873504859184,0.9916127624491993,-1.136623162181129,-0.2577801235663723,0.2010501017874122,-0.6383979039028312,-0.6700017614465094,-0.2402538301012276,0.7335345265296498,-0.2621902110462099,0.1378938862643657,-0.1964540934184615,0.2786970091803584,0.137500866766691,-0.9731332181669968,-1.022149275827175,-0.2812373783107652,-0.2105834921606573,-0.6113439739789748,0.1341913590551827,0.152550582743428,-0.9565762396388297,-0.09835298702272187,2.098977488978603,-0.41218021207963,-0.1108931733385401,-0.4735726714566665,0.6232147478631747,0.3747785751064764,-0.9764084200480976,0.4851707425255032,-0.419823170977342,-0.01381659571259495,1.745828441132579,1.215146495035693,0.2629918691597272,-0.4262435394675454,-0.3122841019186792,0.4159409314850682,-0.3430103356270292,-1.109240590646653,-0.2177982462884044,1.634845452870439,0.1934007238064456,0.1782607274014968,0.4045124506213337,0.004062164723450887,-0.3714140629274159,-0.2635645782693331,0.5874492004741146,-1.389030898980423,-0.2166191461395419,-0.5841205270309422,-0.01734556386468402,-0.5668052396921068,-0.6568086146409708,-0.3681766913160563,-0.02363174597325647,0.3572483163037518,-0.9443317496678928,0.01585634235660537,0.03914624753043192,-0.01732289361608277,0.8972862566131803,0.4175017685263927,-0.4281413793864182,-0.09796450833843158,-0.4431412496255283,-0.4151428866483439,-0.1954213075842222,0.686980629095777,0.008250397324740701,-1.156984233726678,0.08791981394211176,0.5575599051834434,1.575167049083833,-0.06264505693559491,-1.449224229136178,0.2894351724447674,-0.1265245260757221,0.04894335391384087,0.3784333682048588,0.04413787646240171,-0.1008831824684441,-0.7252573894490726,-0.6943583345370347,0.03480757857837771,-0.3023173917793046,-1.330158957815402,-0.2634526501630526,0.1153832810742031,0.1090989151521514,-0.3824023305843193,0.3403901035709326,0.3942200585300409,-0.1098319131433705,0.2194337807038574,0.1870442079903233,-0.5974230099669861,-0.5050735351426742,-0.3632591973415266,-0.1785955460271293,-0.06963216495475048,0.3428346947565336,0.3810636057783076,-0.1509659004044937,-0.315620072819831,-0.5375750544050132,-0.6910260679265836,-0.0972006381075937,-0.7680609814507255,-0.5116295783543703,0.1374927385582814,0.7792061477278676,-0.178631613793625,-0.2647619217297468,1.115252432147957,-0.3277261229414137,0.4482456193892172,-0.1046353679046826,-0.113438947316678,-0.5591858876044706,-1.474470081273487,0.1787869131415709,-0.504284204196333,0.01952935338766103,-0.5761532167065498,-0.2478265611688634,-0.3516099538827825,-0.0811019463302256,0.07221916662299101,0.228248844882011,-0.1969132307076777,0.6979173051592727,0.01913480225697443,0.4118541507530759,0.2098970039239919,0.9229917794221242,-0.5255757712209694,0.2944106231165315,0.4546533403120503,-1.288332290287157,0.06580221852639906,0.1320567204819036,-0.5599467524301051,-1.09715227667863,-0.0888855464356897,-0.4004636733094146,-0.7156793902510188,0.2212057171217006,-0.7023326289382175,0.5919198954082833,-0.05006291127065342,-0.1568685091269668,-0.1240241279961854,-0.1272647064444474,-0.8304203588850315,0.5051695738067101,-0.3366635421999857,-0.8684366458514466,-0.6789979247781408,0.7228743926041004,0.3199923522398828,-0.2982508666681002,0.1479836101628696,1.264775112981225,0.5732162678045604,-0.6363189683482879,0.4434603447408336,-1.158075212336521,3.077905333201778,-0.3004156671542492,-0.9840784578580301,0.2605604538541075,-0.08786101406985232,-0.7558048180331645,0.1074670343017147,0.05130801365042625,-0.1102398496109427,-0.09104802435117444,-0.1143768549958768,-0.5273188302737185,-0.3569763965581056,-0.3596064495440743,0.6279251019356773,-0.6693614133499304,-0.4676328192517682,-0.3527200070356251,0.5856632752826034,-0.08142417114999316,-0.2360379050416581,-0.6708491292789986,0.03760481435583678,-0.5742573384649312,-0.3785991246163753,-0.2395730348587816,-0.3851125898796935,-0.9048958691786304,-0.3120285822928429,-0.2664546142473019,-0.1166449001007796,-0.7107588782411076,0.1404466853976594,0.1419489055495118,-0.1673784610194384,-0.09554413804065695,-0.4652005999762802,-0.36966768001526,-0.2009344954559215,-0.8392111127609128,-0.1849724976279039,-0.4600043463387581,-0.1017642435523891,-0.7431932994983792,0.3480606794287802,-0.1222202908538038,-0.1484279705207508,-0.3914167696784417,-0.1111530196082896,0.7290464607763647,0.399365183599251,0.6373321310323057,-0.342638740603219,-1.004618808634366,0.08594969154198163,-0.09773223112762083,-0.3553826884216359,-0.01862787598126207,-0.1130601644320286,-0.02773344773181268,-0.1786582444926552,0.07480377131295553,0.2566871260249892,-0.04824071322345445,0.1115820744367383,0.1240268266411933,-0.5271793714992696,0.003902125794222921,0.2797250376611089,-0.4316614968306085,-0.954473805331623,1.523573015754932,-0.3667968308041238,-1.003802093224539,1.902705860354331,-0.6494604803609078,-0.06324212795082766,-0.108967292224817,0.3554466729722863,0.2997858256737538,0.1674867727146451,-0.8364863424072179,-0.4465904628973182,0.1141476015556465,0.5489835726656839,0.2823301182909687,-0.05498432083782452,-0.1121970215820088,0.4083209801423954,0.05931871507880077,-0.2159126881495705,-0.3802391019093387,-0.4497792601500504,-0.1445713767504852,-0.2521917672007311,0.07404589933896551,-0.4971493279050151,0.1876422450501127,-0.6154790866589642,0.8266760636275285,0.522047112415968,-0.5252840487430314,0.5069701247188473,-0.1083359290279497,-0.5660719886843785,-0.7492896189189379,-0.08216857755692954,-0.1942630193924552,-0.2877639901757447,-0.1965590600634104,0.02460283967902905,-0.6347000389719981,0.2818586890907967,0.1400186821721244,0.1062377989827684,0.1560812735702617,0.0081054301661774,0.3130090473794285,0.4412426805247031,0.2398014753247925,-0.1711747961420869,0.1899555844545992,1.309618726355473,-0.1415590118831082,-0.1229827254231219,-0.1035397453384927,-0.289834139006733,-0.3075202529049396,0.5169045886968796,-0.3711755056520269,0.05621941756271562,-0.09100826250655364,-0.0532700523868431,-0.1615338525280262,-0.1895558498647002,-0.3465829130155015,-0.2344200956725337,-0.159365171542009,0.1229689792014575,-0.395809857212305,0.437128247222668,-0.1203721827993364,-0.09576793072968164,0.1552845195464,0.05619401647449075,0.08435764573834545,-1.00195152345992,0.2589303629315204,-0.3320169175322186,-0.4427926183329026,-0.5601642201822982,-0.187090552985098,0.4191578411163988,0.07184838960286759,-0.2720696993577223,0.1236627451269514,-0.4497186536807637,-0.7992471467424823,0.2598483912469287,0.5078332814938339,-0.5494236723116003,-0.2880427993569882,0.3483646535205738,0.1099492012877365,0.7277119394249377,-0.3586093712400044,-0.02028007943507579,-0.4277552864198662,-0.0534717762467389,-0.09168897081566768,0.3945512740569068,0.1017012666080264,-0.2978032682905759,-0.2086288959738427,0.001983578803866543,-0.1166787285807829,-0.4823369121146473,0.2300669299228928,-0.3207781350890008,-0.1329585750329152,-0.4021867506719959,-0.05198528263769218,0.5998634939864113,0.7684709417529317,-0.5384126394935319,0.1043576447557862,-0.00301476231969737,-0.2024087592419822,-0.1644480338135625,-0.3568874592477697,0.006466421158137293,-0.3515844162351948,-1.099142139732279,0.1177672696282208,-0.5758777284977876,-0.08982222144256402,0.5941237290990034,0.01120383587388191,-0.05179558677643996,0.003979689935848772,-0.03010319609337983,-0.6420492328497179,-0.1047310187472338,-0.1739462136786597,0.461445134804445,-0.181736617946652,-0.4655824868487636,0.1686890175503786,-0.4096595206848825,-0.2841667502046093,-0.5796827307670779,0.9440075767230311,-0.2230163190305065,0.1941958446846516,-0.4798251029705253,-0.4616506604351631,0.1603678023298371,-0.1500355415104767,-0.0523102144619485,1.023204457375957,-0.4937328688440893,0.6295223998917964,-0.6965018493392324,-0.4029251828836607,-0.6026858803098001,-0.002802695343025848,-0.07280956572204705,-0.2700359393657626,-0.1812306569485127,-0.7154523328827714,-0.555025912708045,0.4654945888683022,-0.9061138502583922,-0.5250669446316857,-0.6963652549960441,-0.3985754935922696,-0.8700342201673091,-1.102641503069162,0.9308331323638341,-0.7901462929540366,-0.4390392951132327,1.004201545287147,0.07245874313867254,0.5246924205521604,-0.848952930932188,-0.3332456540019668,-0.4386870384861881,0.3446632631728292,-0.4153617387085014,0.192827376953377,0.1724054305166674,-0.4093284915111228,1.400299143841514,0.4393412027015753,0.3420752887757998,0.2922960765501853,-0.5041572216614703,0.6287999957413393,-0.5524916447098008,-0.3708587124016228,-0.2211476865274827,-0.02456229682777672,0.1755482996370182,0.5579570734866155,0.3104937323268142,-0.258092486106567,-0.02981864379627702,-0.503418998494489,-0.3219574508812163,0.3229318513306805,-0.307967481325451,0.05946521105872619,-0.1493710421150511,0.09788124131232413,0.1793607945771095,0.2846092108713372,-0.01205022747286413,0.9579521164492626,0.1609706462692606,0.002921904918951937,-0.9164684202111748,-0.1503108985342425,-0.1346231712698582,-0.24816114616396,-0.2531123599627262,-0.1153571492375244,-0.1433571533291338,-0.1669284720587221,-0.002160866130704993,0.7496681739580906,-0.588525082299032,-0.5914601973740276,-0.1412424573199801,0.133157474015059,-0.3179276897868112,0.1917512264038258,-0.05571165706093432,0.2927280318840768,-0.5177830157156925,-0.8312911026471724,-0.08971651439009853,-0.6006931595916449,-0.1861186365876409,0.2567379874305571,-0.1591634720458366,0.3556246945628517,-1.533649959770876,0.05504575466364342,-0.111985106418355,0.1414211279382609,-0.898490079584411,-0.3924812385838329,-0.02041620666322723,-0.4289818006103501,-0.2354209133319884,0.8694207007038693,-0.1771608978983088,0.4103567910521255,-0.3338711141958378,-0.1500948951485965,-0.1329142302545178,0.0466268025909913,-0.4330774766397105,-1.583882332373043,-0.07852132255746866,-0.08924028141368201,-0.8467003183951697,0.7021840627147163,0.1933812866662216,-0.05150293071298497,-0.2507211377363889,-0.7990542946652726,-0.5326992140231042,-0.008620347750403129,-0.5342192162808628,0.2930672089560984,0.05022496609645966,-0.2150511172634727,0.8140585182018339,1.607485304199622,-0.2087345573651458,-0.3222954878565858,-0.4222705433814554,-0.1560147809825445,-0.5706056980973929,0.0424857898150969,1.466735533170125,-0.03525283401042246,-0.1308251174300277,-0.1990500178279759,-0.5826645316960316,-0.789460198101915,-0.7011439201798251,-0.3036207780240534,1.081794134189318,-0.2517180004815845,-0.2080456348065023,0.9800453912525361,-0.1275472882282616,0.1666490686037209,-1.116671399827699,-0.471253738136284,0.03178349826240509,-1.320910473069129,-0.4026863118353615,-0.6721719626215762,0.2794245641060812,-0.725425851589273,0.291414522679801,0.009406068605816614,0.0521292028721901,-0.1796030474068336,-1.212437518320097,0.8309713891439227,-0.1714481393291923,-0.06003898622422311,-1.010798134843914,0.1154458445195204,0.3806623018430255,0.6615528242808912,0.2935349566516153,-0.126784148115684,-0.1948355617691301,-0.1468862613398512,-0.2365584876889325,0.07243810742904098,0.06327572380199714,0.02549457371148014,0.03414744779446278,-0.1896350107909791,-0.8350969284567095,0.5395155646150361,-0.3742466050879499,-0.5498277548336752,-0.5861839716791603,-0.1098452935494754,0.3721918104776833,0.08843461116918962,-0.07159419372088829,-0.9115547626495538,-0.7966785560113284,-0.8547207677805475,0.1281594392249876,-0.5368963143228039,-0.8912194812639229,0.800533746734618,-0.3227957964202011,0.148441428013748,0.4015502981937775,0.1335746678195356,-0.6071824808824079,-0.2771805391128428,-0.5948055780459784,-1.005093888320009,1.399595855439582,-0.7376534203236322,-0.3680221865240767,-0.06568256309744928,0.6808739694236677,0.152905989989491,-0.8447118489326573,0.4305621496422395,0.2913704851134756,0.07942563331984023,0.2076689112136074,-0.1403806202812734,0.7728017577671751,-1.141287470557204,-0.04443851610096432,-0.4207953092083077,-0.5956914727623916,1.093144048649382,-0.3231292917449582,-0.2085818578902996,-0.2008746624818525,0.06643699022159295,0.4549056791799542,-1.079772431886848,-0.6053602526080962,0.1906453486791544,0.1683991759320664,-0.06305522174211968,-0.06056105406411278,0.4496584239636602,0.01439728919509683,0.07563161451050295,-0.2370684192075233,-0.2631695935981086,-0.3178480988427428,0.2244509299369173,0.462734568997958,-0.3287655906240139,-0.4368794296582725,-0.3539104435160862,0.06273196184171415,-0.3019900940853481,-0.9213342568644635,-0.2226720056980946,-0.4270173311725886,0.4139057363221186,-0.7975606077350893,-0.04834380594360944,0.3563451012942866,0.03929389652584259,0.8491590510718088,-0.5615010016801261,-1.94908973514094,0.1067449664273531,-0.7506000675799069,-0.3547378088384049,-0.1680168280643773,-0.3144209462180096,-0.5047003076465517,1.563160651304685,0.2068871111083297,-0.07102616470563196,-0.1292664191408097,1.44691606976415,-0.5434785598308548,0.06638433208709879,0.1529132790749265,-0.2226235736784291,-0.4922750460876361,-0.1659274002506675,-0.5345816864713152,-0.5908746327882047,0.3478517402377668,-0.7869891384923094,-0.3167135270754993,-0.6955467678870669,-0.2748079990746716,-1.169259779424562,-0.1508499142702034,0.6791146382858682,0.2978280034714738,-1.866076520074295,-0.3341545722182085,-0.02974964289733125,-0.660609602469312,0.1553110836942486,0.6293204913182011,-0.5407784122938768,0.2568963093712649,-0.1600118317405697,-0.2113967097023274,0.9069261361279387,0.6147315312085859,0.5381174904584971,-0.9654568483357496,-0.5796394249175522,0.1300151494309222,-0.4718862871903824,-0.06993081187943963,0.6472852077756708,0.3327282209506511,-0.0341187291405592,0.4358219531905851,0.005839239992924791,0.05332732418207487,-0.9581742079325383,-0.07946156710819278,-0.1087582826445937,0.6447748518819161,-1.106616159078721,0.7326971909792914,-0.7169148541294528,-0.2040782221322277,-0.198170749957907,-0.7429906816392751,-0.3231827225588825,0.009304865801427704,-0.1179437464652277,-0.9502196716916224,-0.7495502567644856,0.1686556697836938,-0.3220787360977113,-0.7130805406243048,0.1454922709150224,-0.4857919218750836,-0.145533140407513,-0.124898526103429,0.07921987243984938,0.2990879814477087,-0.4669715965681974,-0.02793125824058388,-0.9760436935370733,0.569838438726528,-0.1520337969235691,-0.342652717559345,-0.1647298920969707,-0.1974559249425422,-0.2306647130125496,-0.6213853813145923,0.0698664262488244,-0.2086349978962557,2.444534886646239,-0.2528638691905061,0.08270495649398667,0.1713914434703241,-1.317459304566821,0.8096751959513034,-0.4710148663712035,0.3381701764287893,-0.5143248225195616,-0.09646630787937954,-0.4249023584223535,-0.4463548742145048,-0.2307230325586903,0.008672126213823083,0.3113411421750658,-0.1653396002883355,-0.2670255969206612,-0.2477573557508623,-0.7056338218137805,-0.1003273925046331,0.05329337155372047,-0.08343284013624742,-1.351780020954875,-0.1115477703443847,-0.07708800707839178,0.2676076948961599,0.3941971866858926,-0.593959756683931,-0.482372236902801,-0.04586244500865976,0.1558663945934257,-0.1581447316432048,0.004701097814435748,-0.07345874886270908,-0.5346522607152255,1.274611462343412,-0.09245075629433179,-0.09826025804966089,-0.3851211209733977,-0.4823096413204527,-0.2934389208937844,-0.0252492239128693,-0.3975962948991458,1.188050122664584,1.614781333416277,-0.05339987745004613,0.220041750556712,-0.5517642613441162,-0.1561596386682555,-0.6147726728604626,0.53397914406304,-0.1259243608530765,-1.220779716460169,-0.0896835580873311,-0.3969241676053272,0.8438384241418104,-1.387995030989613,0.2097603080106855,0.2844551196548976,-0.00744587933717268,-0.2401726867274893,0.6867107035901309,0.2924602720435652,0.1882680696998694,0.004013423718164729,-0.4164673547316682,-0.1086896414235415,2.580651240865929,0.3751964929497534,-0.7664939670274679,-0.3368482078050158,-0.7152509506570196,-0.3171030885774555,-0.008173680607761448,-0.8098122621492874,-0.07960636297120802,0.2283130659489393,0.1514437831150274,0.1224805624768384,1.285745611599344,-0.124686536568107,-0.1797326503248023,-0.728327674622072,-1.036229844744931,0.02906158460200008,-1.037456485218914,-0.1766415771551076,0.5857055645711171,-0.8762817463067207,-0.03682096739918855,1.365735328138642,0.6863879520471399,0.147706607340059,0.5258111491170728,-0.1669005712576691,0.4421741636691859,0.22256221955032,2.258463598270861,-0.2768314397983114,-0.2857004505905875,-0.5053173244112548,-0.3219822944943643,-0.6805731439947574,-0.3632399575975274,-0.5647349195433574,-0.1041139362250022,-0.3857244692707417,-1.192359020521728,-0.7413788081198683,0.3716875700290234,-0.5376495744424974,-0.307413901810596,-0.1077891553523585,-0.648131941574418,0.02897729183993953,-0.06548044237087296,-0.7836495828997674,0.7716907597583376,-0.288091769023777,0.2840007655853566,0.1238911667381849,-0.2383474179862964,-0.1191659893762478,-0.1464100387761762,0.8422636238565228,0.5182760433653024,-0.0259056404199982,-1.62130080318422,-0.4246417063757751,0.8010900428163199,-0.3868002659496818,-0.1977389562838033,0.7101491517691579,0.07491201742694995,-0.2236425178388181,-0.05666280981254413,-0.3753443852785416,3.384828381764038,0.1343564893491984,0.2183870359104053,-0.9637688939963213,-0.2214140323336053,-0.4920751680683118,-0.2227577793299906,0.3882213076534278,-0.609852257240883,1.061833069485253,-0.4905453590894055,-0.07138696249576207,0.008737638214694496,-0.2821774223914102,0.2148988674843509,-0.2117322301734627,-0.984336893334138,0.7070366924426542,-0.1589849380676583,-0.1147002152732392,0.09526518789408001,-0.612916761181304,1.134488358722985,0.3410486704845251,1.902079548797167,0.02930179624851365,-0.2074546729052145,0.3118032181184549,0.5052286438775604,-0.412751461201817,-1.190906635084773,-1.088076655709083,-0.4858629982166246,1.055428236046636,0.2617106002474011,0.5761665741885613,-0.5306903236420618,0.1719156859417699,-0.491112331108844,0.432412233496949,0.01779325370695815,0.493553060481573,0.4103770623832357,0.01127721324031525,0.0857373496476488,-0.1700924941659133,0.005363726318708105,-0.4193995020436835,-0.1718748379420252,-0.1697002658506754,0.2176971636488301,-0.1490209922713217,0.7937763509532694,-0.4600301459505551,-0.9019678593240357,0.7878715416694041,-1.067120889624802,0.01045816786759852,-0.4657183500594958,1.124859000346744,-0.102379797791123,0.1251605838312304,-0.2363817834304307,-0.389206514281293,-0.9326966915175746,-0.2264704881299629,-0.4789059197421132,-1.130004755666949,-0.1456220733949325,0.08757353352439023,-0.2697514566737302,0.722883080619691,-0.1030819712106665,0.09639579507250295,0.5433451224701814,-0.2422863787088927,-0.2326207244789653,-0.2628973519991994,-0.3130578040806234,-0.1157412910666668,-0.2763965588432514,-1.364446603430255,-1.252346882224965,-0.9166534979034128,0.07926932080675228,-0.2978560127155795,-0.4274213339704263,-0.06330059852001642,0.2161796687332437,0.02236965023139462,0.008801969442827906,-0.4392348511331137,-0.8143181611944982,-0.5048122142526345,0.3778152150041751,0.02464447931835704,-0.5281164901695611,0.09135255617818004,-0.7662577437767476,-0.3592119742911277,1.425514195423447,0.07367111125883516,-1.098636546097749,-0.4164183227273441,-0.5645118165617522,3.050007634056169,-0.8139512390395801,0.05381540741379229,-0.8091617281143312,0.1291712011622982,0.04726910626444285,-0.2104948564358969,-0.4854453379324364,-0.2109620709043327,-0.1545093248138181,-0.5345798690646785,0.6749975465249705,0.5721343096114541,-1.280093864509424,0.05866702264347087,-0.8845245189540166,0.4817232213501767,-0.005862634336513915,-0.2724592729109793,-0.2867450516369471,-0.3797488955116289,0.4741449661179598,-0.5078957244892851,-0.755265412850358,0.09896571074701654,-0.044096800864884,-0.1191801942906527,0.3330956310770584,0.04799115193063964,-0.1695634038225811,-0.1513400182328341,0.2369669801300398,-0.2274780842290237,-0.4992919018587401,-0.2664915684831968,-0.6923239873448275,0.1530735632094335,-0.2270802716303325,-0.05937930033125514,-0.2050870511562523,0.3466249442875756,0.3306561968951041,-0.1928339217710293,-0.8470984655494668,1.294271060419886,0.0006592837398840815,-0.4966946226311523,-1.072291038642266,-0.4582206022716583,-0.2643947913567617,0.1823535330248281,0.1837927351987877,-0.7066759262569118,-0.1004424450046521,-1.202992449219475,-0.2726403786772992,0.4187606053732406,1.360581678559742,-0.09536759153399868,-0.2721255587725275,-0.0503154888514189,2.252359687549911,0.9471573598802184,0.4409188164018484,0.3872316534658769,0.5125282204946503,-0.05792467389776926,-0.6015696330926567,-0.383119695016707,-0.6675450710259992,0.09820510792400232,-0.7926063844184614,0.4679431466550023,0.05315911038536292,-0.3873489872009412,-0.5306393713422595,0.7846937425408599,-1.13750157903408,-1.503055995873477,-0.9792187023363819,0.2251571117403378,0.09781898746676133,-0.949649698543943,-0.1999076821076623,-0.4099396464959784,0.5264658949228377,-1.024247731473634,-0.6705468400962952,0.244873170817767,0.2111427590286809,0.579272782147863,-0.195333509469872,-0.3560638407012189,-0.3118973884645737,-0.4209715090779324,-0.3336261701723565,-0.2998862430467947,-0.5808043534423117,1.154767799972549,-0.2446942302728965,-0.2709380644260605,-0.6241314741644391,-0.4803405241622244,0.1456300129573756,-0.3067834551334188,-1.268261345821297,-0.5293742219667273,-0.8528958625666559,-0.5276061947961463,-0.7928498075285397,-0.3367182295037403,0.0383420270789044,-0.1919535844764003,0.3127492748898786,-0.924784102206883,-0.4696746228037275,-0.8023467304304782,-0.1692113047060773,-0.4701223422758887,-0.366078214935409,-0.008139055237721614,-0.3374807213336073,-0.2733891789250637,0.02564560337608141,-0.5000800589626331,-0.8341223088471061,-0.6509124240798875,-0.1923725679261775,1.931648091831579,0.4082360334598701,-0.8890193818331311,-0.2379154580161974,-0.5351400959303549,-0.9658591410470568,-0.04037299141939117,-0.1809838539569381,-0.1778016386966688,0.1194140094982107,0.2336496760207033,-0.6661353581736285,0.2386307618562436,-0.05745738895773316,-0.4415296156559133,-0.7069010872272314,0.8832653793264498,-0.043956037905813,-0.7610447395661906,0.5057437782785624,-0.3746982150430833,3.812850820824713,-0.3831175833892024,-0.7420698146558463,-0.137999112935552,-0.1762375309168203,-0.5279646559509646,1.847244331941997,-0.5984388402224958,-0.4079089260799146,-0.1728255701170925,-0.6064827810535395,-0.3160975227516703,0.9576012647798585,-0.3436149219925603,-0.04543026598993589,-0.4049769333057942,-0.7720487082309715,-0.728822508457866,0.05764470577290597,-0.3421663334673371,0.1474931938188359,0.3822795044800424,-0.05981850129479625,0.4324921892796864,0.1511917505168126,-0.4546266388564623,0.580200857526007,0.2876301263476349,-0.0912955011772949,0.1258597237114223,-0.195193916658955,0.3658905648828359,0.8105186654209015,-0.05509681686346458,-0.2169415520863585,-0.5163585140975256,0.4699921754410077,0.04402098731561663,0.07089078378952723,-0.3299306255143478,-0.7038504795227469,-0.6831203772073698,-0.7145795149893284,-0.620907378286087,-0.8906470139690988,-0.7586934802147317,0.07515288983746375,-0.7767764973355863,-0.1644482930001326,0.008546011665575363,-0.7034767562663982,-1.154324047253683,0.6584879956028815,0.2442687084685009,-1.722744325363792,0.3283131932195679,0.528058262567536,0.03579545149914218,0.1286536341382143,-1.133718428899421,0.2055010654405653,-0.2970168111182093,-0.03336500787521375,-0.1611333809757824,-1.00962535504787,1.125891681046505,0.202664851230718,0.3955251476843821,0.1604105230626962,-0.1198479971771474,-0.5756715913412784,-0.1118339211325755,-0.3177472863095413,-0.2363739719232483,-0.3834486089470171,0.08503594697546951,0.02292761279561704,-0.9279788532395405,0.953075966631453,-1.005700877787488,0.7834530112309028,0.07392152486199664,-0.2228057719711925,-0.683246789346078,-0.9010667663466987,-1.10884029139616,-0.1577408175722017,-0.06972047028355657,0.4130320442905716,0.5569510343159213,1.666333240667712,-0.03293894427406374,-0.791150961508825,-0.730472821102925,-0.573848382033834,0.8131345741244851,-0.141670974008373,-0.3343939633955244,0.1395358834637312,1.777892933693361,-0.4584902942793221,2.164088053181109,0.03695788128773241,0.7859599079442943,-0.08958428583132395,1.340094258038871,0.1479576523025332,-1.196597908666188,-0.3521215439080881,0.4200339948759193,-0.2255101450531204,0.2613873919627588,-0.4698379364454734,-0.3291778454930038,-0.08720812053524507,-0.2165599599995117,-0.1424494627906155,1.790536198838547,-0.4329366977807069,1.005765513485742,0.1333511246398756,-0.2171604630713466,-0.9832080628449088,0.5765394700501711,0.3717840843927743,-0.1527976649757329,-0.05481973658204811,0.673979492183585,0.2382618195956293,-0.6797103227758728,-0.1253963451380048,-1.338703985696223,-0.3988175029147196,-0.5249213881696075,-0.2032702834302361,-0.01339927073792113,0.05519061881533466,-0.04775410244018014,-0.7088907707537763,0.1779932063905037,-1.093322328990242,-0.1480766473099135,-0.1157601198758453,-0.9457498552950883,-0.9874915372267025,-0.3086503066045063,0.4803164804168147,0.1278288521566898,-0.07373785460397635,-0.2155173395611311,-1.174808517319152,-0.7168357303084865,0.7135508491731899,0.4042936805547862,-0.2963750193008252,-0.3367191100859259,-0.2601903123006786,0.02397117666830688,-0.09166163718190963,0.1009603738495274,-0.3085688258524865,-0.4110457400380555,0.1412226730124242,0.07647220085921223,-0.4255634465773459,0.08957526335078857,0.6767428865430424,0.1866117694956443,-1.013596186873963,0.9802983730127302,-0.03500660505039545,-0.3441443537396466,0.3175038125074904,-1.258751484091715,-0.5087334537896909,-0.3880716344163235,-1.362818295583784,-0.6592873032302635,2.349909240430315,0.1856809585157456,-0.1318448290412963,-0.5116426278610441,-0.2717441531364848,-0.387531951794318,-0.5477555582234195,-0.5920313549024255,0.6111216342558394,0.13750389949755,0.6216283249105803,0.9104602953204634,-0.1965348432477693,-0.8301710792356333,-0.1163263615182816,-0.565240549056116,-0.6978897591265423,-0.04513397887892448,0.1325626057895914,-0.328415223436247,-0.04266006361576686,-0.3045793983819144,-0.02806462181614573,0.1418005440735776,0.1608758387221279,-0.08828893635557172,0.8187663567960139,0.1691694793647882,-0.2039047972722245,-0.5741969146935934,-1.105909157095151,-0.182385239747503,0.1518564232648789,0.4219599178858856,-0.3384874904115062,-0.08336782393222446,-0.7331454207030289,0.1007583866225791,0.8338328023885817,-0.06441723338917932,-0.1574768068291911,0.01573853687893276,-0.755691019972468,0.2143585622113509,0.1886069245283042,0.05126359021368628,-0.04471801948940524,-0.1848651608380624,-0.6944424790033676,-0.3933533129964983,0.5078621824639604,-0.1296264879031035,-0.733477908973718,-0.5958889346304079,-0.04515250811774273,-0.7150718428122358,-0.5442550337485802,-0.9925478012772694,0.5544175334352799,-0.3692099569015422,0.1395514754104281,-0.2881968468460068,-0.9021987709483591,-0.3404017080725376,-0.4089062359353797,0.2476177888059314,-0.08689416642989498,0.1767949516266138,0.05081921144400261,-0.2980857595195252,-0.06150933163870755,-0.8266674045341883,-0.1890751312827919,-0.7890813908209803,-0.5228071748408034,0.03243243310319853,0.02019303591532972,-0.06647128717316451,0.9025970472285206,-0.2424953521289973,-0.05127198892153181,0.4264679116829084,-0.6658669225784513,-0.2917887094067543,0.2390437279728982,-0.1534703223065224,0.3439430349792871,-0.1487708968210157,-0.7416212302415152,-0.2682517997466923,0.3197465934993121,-0.06888734598712169,1.143640474926783,-0.8413050363682659,-0.2512364992785859,-0.1124317636658628,0.8728716930108423,0.4776880338822862,-1.11257452133877,-0.9897810392747168,0.7422279724870775,0.2116177244994193,-0.5738321292752655,-0.1002974868201784,-0.2132293178443387,-0.2639874830874034,-1.983866568508293,-0.8067316275729773,-0.0893240124438326,-0.5700980226462018,0.3401987943182202,0.4806396101095006,-0.3037741097365875,0.2178273102665764,0.05470804787314654,-0.3229414989754875,0.7257398579004011,-0.2235357641414238,0.07961825879720269,-0.4683417491499453,0.2234540407737668,0.8473367748020054,-0.0387574934364107,0.1674515345418505,-0.72532152403628,-0.3615246506855835,0.1494686205069423,0.002071065629639305,0.02569459547051364,-0.17744084661869,-0.115765497456273,-0.6210156941426,-1.322866748197097,0.1233984219439074,-0.2786700944369163,-0.8572113888231898,-0.43312972378882,-0.2387212615237857,-0.005538021932747021,-0.3535171109694783,2.144049393506659,-0.3844407330378678,1.144325100045648,0.2135947607733978,-0.4321549338297873,-0.9119871104889069,-0.35847443229916,0.2909643192488168,-0.3753317450029767,1.127632323071969,-0.1834007858941065,-0.1604099757105255,0.08652876128511224,-0.05999451338088862,0.254681213265675,0.5928260494497358,-1.023247269490941,-0.08503273918755318,0.5897226041883984,-0.5694153330082526,-0.718677679476212,-0.6098312164669383,0.6979781728823122,0.6836900754875868,-1.19503115384848,-0.2996666591936033,-0.1264170286769495,-0.2868361386024479,-0.09363612815560537,-0.550501449394637,0.06467444279046981,-0.8226024622332082,0.2088431975191914,-0.5007898223283783,-0.3299395229002672,-0.2583036125990036,-0.1339004105759338,-0.03365554947369549,-0.5437410894586933,-0.3427838891873421,-0.4657065110593536,-0.009611318693043436,-0.4887107966191281,-0.5479912099074146,-0.1865268407459681,-1.182881819984099,-0.9936484274905223,0.2439910590586372,0.07186705930078865,-0.420505850735818,-0.309101987991554,0.07416301843242291,0.04141777011191368,-0.7360687536246723,-0.4698508303862329,0.4869978354888006,-0.5005978740357983,-0.132248551132953,-0.420323143248742,0.34862435179547,-0.3253636204530049,0.08486657257164014,0.1416698240961877,-0.3973946880004324,0.3324318710359591,-0.7687105891841801,-0.6367072712169787,-0.2890107953670938,-0.347932248821993,-0.0142317750641677,-1.751840585913052,-0.0705318212867643,-0.4969744659114931,0.185462263632906,-0.3455699164945322,0.02767544360358806,-0.3914993200870308,-0.3797126935047773,0.03937472434544686,-0.2632672995689284,0.05845738197516506,-0.2378233470867597,-0.002403803152632165,0.8671803193975242,-0.2979881950228909,-0.2086082299725808,0.09805388779155642,-0.489646662741088,-0.1158400097653701,-0.7253070264492891,0.5459047152807721,0.913335946389648,-0.5686537026690239,-0.3207638266400901,-0.2123118903880681,-0.5254965984267307,1.703805996321849,-0.4466104469262432,-0.5388546200576688,-0.1639620591309264,-0.5698043315747627,-0.07018986439860897,-0.2134818185339845,-0.3020875650661047,-0.4140991528386392,-0.1257731166547846,0.4762079165932547,0.1981577571737072,0.1460472275414916,1.152950696971952,0.07486070990437964,0.05059688325046163,0.8061911102366704,0.1903137322570516,-0.2832218127575433,-0.20273503777951,1.464131692562479,-0.1752374833578869,-0.03853836277043599,0.2187410030700042,-0.182915893995022,-0.01270113812750586,-0.6642096476157393,0.2482007350466547,-1.414404333989275,1.26454447532555,1.140674951886693,-0.2274251467102341,-0.1719223914809019,0.8061543326533709,-0.1958674438232095,0.042196900929026,-0.4102599971336197,-0.7877668682416038,-0.3110283087498699,-0.1911177855532846,0.2609963602565756,0.1288062575432505,0.04939579113548544,-0.3513542586322878,0.199180554300042,0.5579899407568218,0.4498918853450835,0.6141808104397659,-0.07808309748362141,-0.5765649620255811,-0.301474506033416,0.11258104576373,0.9527833503394968,-0.1270660858928815,-0.3815679339592872,0.2285512798064738,-0.2407735392855296,-0.2856658212317831,-0.443968988425712,-0.4817552253898648,0.119041160693574,0.1787327276119831,-0.3801308672314391,0.3437550338608901,0.3824298827183878,0.4683399736124557,0.1719158605528186,-0.1595291390305524,-0.1484666322318575,-0.3594310513878508,0.5996480422117948,-0.2453281098692021,-0.05937564161428071,0.2747316803292384,-0.02094700570856091,-0.251762811975485,-0.1062832517664944,-0.9189133536989218,-0.1452458645907435,-0.6070856239745259,0.5153198559223794,-0.1623933542243791,-0.5798393748883021,0.4971279438206333,0.8096101137461376,0.1052726254731558,0.05403212215434863,-0.2170537705669525,0.0856221084809516,-0.7019597182027135,-0.4898809446180941,0.6711183742903204,-0.28286636767867,0.3182255407430523,-0.5269394210715076,0.04391801958437296,0.145246839184282,0.06991218886194901,0.81117324773008,-0.4712245314553429,0.005180055102888519,1.720974242283351,-0.05031048637299797,0.0901169637716709,-0.4884301457881124,-1.053030522097803,-0.2635942303721344,-0.1783389714640495,-0.08521093335797866,0.4784281077299125,-0.4542693778837215,0.02456365257155812,-0.5202503834235891,0.2126372094403676,0.5464767544025899,0.2708131287813185,0.8089174897727992,0.718125792599876,-0.1333552239600885,-0.8668154865265164,0.1001939110836561,1.835114992597453,-0.218181540502871,0.0128713558395117,-0.6174702288461652,-0.5177598805649583,0.5373536843996192,-0.05915101498783122,-0.5720320239000085,0.07465422213987809,-0.06862210313515552,0.2858943074167283,0.5298814186077644,-0.01768013334296873,-0.1959808254489918,-0.1427890030869952,0.5926278046638102,-0.2733755283934667,-0.1288019460970765,-0.2718684423480088,0.2139308167108956,-0.08887621954846958,-0.03909395655021348,0.3520162337111621,-0.06411687526608276,-0.3303516256477383,-0.126887537673313,-0.4818394145911332,0.6987185334209147,-0.2011940124745226,-0.5980405363738051,-0.2284463351257396,-0.8149508730262393,0.2136054145431904,0.1904486097768348,0.03234599535602532,0.1071177990924191,0.008575424908404452,-0.1411889123680273,1.139778092995496,0.06704225603927287,0.8742622567415602,0.275173886917404,-0.7878861864048289,-0.1101749291487917,-0.3196020744507541,0.02576949148306654,-0.2511371331337816,-0.6751124648256563,-0.4571809052942366,-0.1998283484695464,-0.1612782681623943,0.3516522515742969,0.9211563082192737,0.5015634462891427,0.1552617938512069,-0.4781441398731146,-0.6043809299730368,-0.04899082122569775,-1.543774520512506,-0.4041502424039917,-0.4080034704948249,-0.2469319646636641,-0.5578201339768017,-0.4627385029347574,-0.02639964472970558,0.700798157510046,-0.1636292285784777,1.357716702587964,0.1711327019389535,-0.7802279529783165,-0.177666560947539,-0.7914227811668213,-0.1103304677688025,-0.2423836468596491,0.43708045456534,0.740932636595263,-0.3510289989850015,0.4373514354307224,-0.2562939996083722,2.007668087723649,-0.03486798761610951,-1.227139902366269,-0.1949762264457475,-0.8075482821622032,-1.045236810968525,-2.519507777536888,-0.6698198721999419,-0.1040173985779652,0.1765033528541496,0.003457774691370887,-0.1224660986408624,-0.2287148981247276,-0.2698600415758017,0.3988455437632235,0.350235571545182,-1.039727802792927,0.01689546363285461,-0.3226638358947559,-1.192503280456173,0.6732270574465741,0.9544918069889362,-0.0582505473201171,0.3944736297468924,-0.7341470146862694,-0.1860952194374392,-0.1809872363653662,-0.5357185776080364,-0.3769368991627329,-0.306771157529252,-0.1149425748784279,-0.7794864200183992,0.6495666297222877,0.2765393115830576,-0.9442023565858439,0.01348568510904203,-1.18676920535688,-0.4525220661167981,0.06297724309477001,0.1031518014022548,-0.04444479161821374,-0.1835475032759026,0.1375741363214174,1.335290605962921,-0.1719691640459192,-0.5012165935244218,0.4882326100604348,0.09362948640678856,-0.03353547753494604,-1.049425723804173,-0.007835589424225846,-0.4903798418736748,0.8466254111358124,-0.3117185806827816,0.1287156374167363,-0.1993447192489667,0.438923498921592,-0.4334319667735016,0.04381104042449143,0.2958335361247487,-0.144063383167632,0.3003389373853999,-0.02419956258346796,-0.9272421742827909,-0.2528690760688792,-0.3095203737932676,-0.7916251889127384,-0.1794341658837852,-0.4202028666669145,-0.308521156150684,0.4289389504984743,0.1673596670852213,-0.3879527964666727,-0.8609035690019065,0.01236047477236432,0.548196468885095,-0.1512475204001758,0.3025486753441868,-0.8623417960160814,-0.1854670915859158,-0.1329178991403661,1.65151142533931,0.104790288664823,-0.5336456211567778,-0.6545137776947171,-0.6850911041454982,-0.4227196009179467,-0.2341075244975976,0.9974931006112403,0.09991394587265687,-0.4825528340428777,0.002433643930225882,0.2711667857381426,-0.3098842394671772,-0.07296591123005236,-0.2152405173976524,-0.4304186337311759,-0.55033543221488,0.04533225107738519,-0.9819788857240765,0.1805914735811144,-0.01516237556236368,-0.3569298546844403,-0.5996902835393458,-0.604745177139467,0.3013689179961659,-0.07653082776107281,-1.534635766484159,0.08430225202085104,-0.2832692189797495,-0.3917614355233139,0.07206023050862795,0.2764767312902483,-0.4812976440909001,-0.7060918358296079,-1.023192633060507,0.3703059937202178,-0.3989254089296281,-0.4648906798080961,-0.6156010216354394,-0.1453266138108338,-0.2731101080292824,0.9040495461277712,-0.566539085679519,-0.2300763519474691,-0.5447695143891048,0.08320475665431029,-0.02344298741101597,-0.1003186453713649,-0.4928372111060526,-0.2600862449816078,0.9855629239162225,0.1436910736356189,0.4626936394317997,-0.4358933667342749,-0.218880770072617,-0.112713815465785,0.002916272232301131,-0.3686670648927544,-0.06672843402110534,-0.4284801608594686,0.1801661559396346,0.795275149347205,-0.1511394224769521,-0.2371623221465604,-0.3852005180897747,-0.6377372113969555,-0.09307887671244652,0.2951939685945207,-0.3761040664926075,0.7595009808577736,0.007967489695085357,0.0767256497520791,0.0142773143939098,0.1231984775728298,-0.08636341833542412,0.2532943676091157,-0.04325241656036139,0.05949387605780411,0.7049149688126032,0.117887884743158,-0.4626543306300462,-0.4331097227678168,-0.5606197682902823,0.04556836045968696,-0.1005081400668954,-0.2663351002821904,-0.003967338556693647,-0.09797441283575416,-0.3294644799170933,-0.08532238351388885,-0.4018880436792603,0.4943636353186913,-0.5197111917598098,-0.179176895113956,-1.07659475337197,1.449343339861698,-0.1203423239563325,0.02487386609224132,1.099531444623049,-0.5797647290802515,-0.276070434171457,0.4486121618420558,-0.6056287512377768,0.1189285739274905,-0.1697821119998107,-0.07293885496145654,0.6840433138995111,-0.002804951538606791,-0.2391757935308242,0.08604462646548776,-0.4041433562823934,-1.036973318941763,-0.2829067685905567,0.01822696147783245,-0.3402143681110325,0.006286068052606466,-0.8460626384540708,-0.1989574302220315,0.3396410896613321,0.5655079857376776,-0.4028792613111243,-0.03750738765102138,-0.4911738300985793,1.191645738353349,-0.8372830910894502,-0.3860730120582888,-0.2647325341123198,0.06795484410722488,-0.9406546432835492,-0.3118186831698974,-0.1578168130643775,-0.08146975816242545,-0.2608203271745055,-0.4619812448256104,-0.01806674161325119,-0.5425390527578148,-0.5588831694299453,1.241262586229489,-0.2752179083519321,-0.3813017961712409,-0.2940502999426063,-0.5594502043843781,-0.1439674847364013,-1.41627920825739,-0.4159618446174924,-0.2286103436910948,0.06153352073198259,-0.7548988843685258,-0.1718555042657051,0.1120984333331431,-0.3995500696437176,-0.8266978042095743,0.1873419713009523,-0.1571896671379104,-0.3035824014281972,-0.09729367351110761,-0.4087536703969538,-0.1121040095657326,-1.384321580985687,-0.08083912850922029,-0.3007453038992296,-0.05011612288029101,0.8838402867124899,-0.4643372628179427,-0.16617943155956,0.1808554580554158,0.2455471562311319,-0.3569797364609022,-0.4317063348467886,-0.6759507327249415,0.4207412476686662,0.2571562700714612,-1.029603824070846,-0.4539964310449058,-0.102586384211538,-0.1828916792323324,0.5111668938564626,-0.6875683031459116,-0.6329042592145115,0.154156556375306,0.1636983057189907,-0.1947214104367246,1.092714777205032,-0.188742302943486,0.307694976371986,0.3699406713585164,-0.4913074901239868,0.02758896301779656,-0.3953384151979719,-0.06704484524288865,0.5094475018554737,0.1495220542418152,-0.3386922124048815,0.8001603910863623,0.0378513148008312,-0.54756843286074,0.1587375881982334,1.779561604090862,1.958209513480609,-0.2755456332169935,-0.01986414183913987,0.4741010970183461,2.448004996663865,-0.131727747349584,-0.2856526769063301,0.1260451273496307,0.151774852285767,-0.02511092910509071,0.4119392442008757,-0.4868898893499526,0.1204892465906346,-0.6278463677346295,0.6400064586374138,0.06922065813594508,0.3929903728078802,-1.376724763035342,1.670700535927158,0.1062964950441645,-0.2101194133053431,-0.3152539402749003,0.3551209028148504,-0.4448053856859044,0.04739479175958528,-0.3440628797124611,-0.555065431736377,-0.06591845096913561,0.5460867804509484,-0.1933081945688245,-0.1535608300622595,1.144039090815877,-0.2603488856789516,-0.2165318944729082,-0.156569514053843,-0.3229486286273291,-0.7325563444380231,-0.7067041581279538,-0.7737472873509877,0.2234088632901042,-0.206559203922852,0.6675975994531159,-0.2981524535087337,-0.2846547436404109,-0.08570313048437893,-0.365659402238348,-0.05943761529990375,0.4584847620176669,-0.0257841682793724,-0.3859756993081904,-0.8067076235299158,-0.7491818331459196,-0.2616065781857508,0.07379678824305672,-0.2359862583391117,-0.02063755285101655,-0.9301066769577792,0.4450481484496768,-0.7175714887942132,0.01498357331035765,-0.3177058942272309,-0.8143050225499494,-1.21535642422119,0.6248497154911916,-0.5214723014391767,-1.117545138126697,-0.02152963512832009,0.216902860559216,-0.1620518864454479,1.04561251090063,0.122279347874459,-0.005812179078537869,-0.2666378098566763,-0.363104713991334,-0.8424175584007648,-0.302438422032458,-0.04872357437191683,-0.8161275425470822,-0.3419703702761298,0.0975050127394457,-1.113352462124901,-0.06904868047065975,-0.2152780995597403,-0.01478178758306475,-0.1822335578496789,0.5465828343265949,-0.8698346331371745,-0.02729402278308833,-0.2063552476950888,-1.491195963998612,0.2420831013782972,1.25752733764451,-0.1035101614254182,-1.351311857211456,-0.8993258853473624,0.1341815024099943,1.297569853578222,-0.3618938276891491,-0.5897347318592985,-0.2460443021315489,1.195554889538344,1.247764817964743,0.4631274647763003,1.195554889538344,0.5482871093273783,-0.3322695843538178,0.05455347831930536,-0.03526403400506824,0.1038916309000735,0.1266824102596116,-0.5601436158621761,-0.05566894932990782,-0.7055034536093859,-1.629358063269269,-0.6178159936373502,-0.9690466786865383,-0.4220186317843049,0.3094186053208745,0.609636718206679,-1.022126406333802,-0.0399817805421661,-0.2664261990515892,-0.1164804784003126,-0.5703211616863681,1.749595513658093,-1.00967438196277,-0.4767416672225846,-0.3417297237423711,-1.000495336037085,-0.0678534411703391,0.1189483854460575,0.2362958087012017,-0.3277449810376764,-0.1191962399589629,-0.2394159060735339,0.5602653791654627,0.8218158181682039,0.4519043620206744,-0.3662944743421769,0.2512702527404597,0.005987035659142871,-0.6684531674748507,0.2640853121531277,-0.3445375917562465,-0.8938169741451921,0.4035528357104284,0.2022500311686813,-0.4091396219920682,0.08171558242764315,-0.2347285958292225,-0.9993940068384639,-0.2278513507099355,-0.6529829052760785,0.2318484865751214,0.4338790200272025,0.694032604062441,-0.9832077349653282,0.9341168892159912,1.83194264130963,-0.2741012360288493,-0.2977569930303692,0.4843786125691107,0.1004196662142478,-0.2742768700426027,0.1624836172912137,0.1303299096781036,-0.4314276947308709,-0.2045435768207062,-0.6269395825799187,-0.7303726711273166,-0.08219883462988233,-0.1239936754338191,-0.5018972061709759,-1.035405434882686,0.100507671204867,0.3703340480110621,-0.03256869465499748,0.08920914321323048,-0.04431643077685816,-0.1488367674336936,-0.1810700129582843,-0.1839274237903956,-0.3782346244739878,-0.5779900406213422,0.2453613508577935,0.8874148552424492,0.9273737904955466,0.05071290245412369,0.02146824660590785,-0.1723689334692706,-0.7112215030300826,1.658689313389789,0.07830895396880183,-0.612325184679268,0.1716639143116477,0.3156048160142703,-0.1108858805626349,-0.3568803497539682,-0.1529926865448939,-0.1001238290710516,0.07150370130752901,0.1633412345194642,-0.2298926470879043,-0.2680767330719045,0.1851125075106586,0.7142685679020957,0.5348467765964245,-0.05451171942178745,-0.2897639011384027,1.444929774107737,-0.4419005632824323,0.09296160447210049,-0.199877191889815,0.9686969421549858,0.1747420085235578,-0.5927144627152083,-0.3889203934610745,0.975605737765308,-0.2124120458941977,-0.5449257199836541,-0.2324899982971116,0.4519215533062504,-0.9220949408064874,0.3826862154788789,-0.2820314239452529,0.9809195050766528,-0.5705420954538398,0.08122661031916439,0.9985225845090796,-0.2489092573327783,-0.09094880502459864,-0.4177227626677155,0.6117930187303997,0.2143578979908392,0.07949264204978372,-0.4047810517920278,1.436333679325863,-0.649548097338568,1.265657460638199,-0.6177687292185093,0.1858561965001863,0.0506261062630366,0.3719415278008476,0.02986233550878465,-0.636467479792447,0.09964731832164787,0.01532822203536399,-0.2644176018830219,-1.107681042618969,-0.2985609687894279,-0.4979059208955701,0.5448909209696878,-0.5607710205618187,-0.6389998965503492,-0.6506901744728637,-0.3720819292332588,0.2566183685058698,-0.4713616549218032,-0.0816772795989306,-0.7036330136109051,-0.2094056695562371,-0.01274378806200115,-0.2529410813373228,-0.2516879423745432,0.7901477041963519,-0.2310625290700708,-0.09986316480740316,-0.3102560539056211,-0.1509837808764079,0.4825688016726608,-0.3529038044861112,0.8653116795234159,-0.3797946475895367,-0.1058016385590504,-0.3385079076121552,-1.24774461023836,-0.2064692095915667,0.04555998593970143,-0.02730943355633213,0.14088390155488,-0.1577536838231691,-0.1259393633952275,-0.3611852279849716,-1.192776528500408,0.01733349530412632,-0.3757954654795636,-0.5037909337704741,-1.046025119662822,-1.093314860634563,0.2291702156594212,-0.2010532316992138,0.6576577154906067,-0.8696388250824512,-1.070226326257434,-0.650601445595711,-0.1313612928257677,-0.4283708543566221,-0.1864133284476852,-0.3897279728382764,-0.1549132147172358,-1.135344551988175,-0.9962901152595763,-0.9905289602300128,0.6412187422164731,-0.1439750309566928,-0.1748299383403964,0.1574060229271501,-0.006418365861313276,-0.07230989111552319,-0.3273095212844943,0.03678361309820351,0.2892700789549937,-0.1463398242646912,-0.2411099654669649,-0.1162167766086222,0.41382329304433,0.2222356720406229,-0.3848185831901999,0.03124807449754123,0.254232957133705,1.806546655775828,0.805954096825833,0.1076450436636545,-0.3506781908963724,0.8504932060901214,-0.5655631085790226,-0.05564261041113579,-0.03546748259749195,-1.123702913990155,-0.2770381903532887,0.639496439716716,-0.7924034501341902,0.2683820749741937,-0.443206199826507,-0.5874578822877086,-0.3133475605837739,0.04628318463737807,-0.5379184221447979,-0.7748865468909998,0.1369780514017228,-0.5993395179595477,-0.0578888112616606,0.6605457933698408,-0.2750722974337949,-0.5918319563221646,0.007068392898872934,0.2205566313049959,-0.3087675982119013,0.2537482705353039,-0.4108395108025449,0.1865613349927715,0.3344780193275508,-0.1977578871777211,-0.1818436645978646,-0.1411790354681242,-0.3613621658422176,-0.4072647484905207,1.284732718462306,-0.4233620528088269,-0.6094940860859379,-0.1890358407667422,0.3566806539929311,0.2892151823143556,-0.1396134314714551,-0.002769491342162742,-0.8161972551336698,-0.4261653951508689,-0.03382459523573658,0.7506795835585219,-0.5819476064576777,-0.6617032502984481,0.2994695501897362,-0.184251566085911,-0.5005996614196246,-0.9675240781224079,-0.2609379460250165,-0.3030669188139435,-2.342138075287977,-0.7253580902862892,1.856620223531796,-1.072777632890503,-0.1440918147686455,-1.020388475632704,0.02293600629043591,-0.8095629466426901,1.114080219783629,-0.4295731079870037,-0.941223547779122,-1.384448306283739,-0.5226431199470302,-0.2065849901147714,0.2031748982388832,-0.1207023465086876,-0.5154509663683179,-0.02792938161323538,0.05423341311567983,-0.2015602567415525,0.3009256684018197,0.4571027935422083,-0.4920719963225407,-0.771588490703534,0.1634737427521031,-0.2914883531580906,0.445307904731725,-1.157866570884528,-0.3693719304828319,-0.03167235349514251,-0.6391727663643832,-0.3831041962332589,-0.8324222745970646,-0.3728917120395013,-0.8048478204598842,-0.3851438551563988,-0.2953215083533647,-0.3918291278238972,-0.1822482219982095,-0.2930982059515658,0.1043396361891276,0.3329070316936894,0.3848329201227186,-0.07512827040850098,-0.4139982411489752,-0.1755388927703473,-0.7302705011359012,-0.4443104664448679,-0.9459510918703513,-0.2943430650580353,-1.170834547281866,-1.165014910488598,-0.5546647193278894,0.1100361798851931,0.2569847498567456,-0.7818000741264107,1.226504701465065,-0.5487857351558075,-0.3762944731288336,-0.7222080027690899,-0.3501050833450846,-0.0598928205289647,0.4223504974164217,0.1374794180004053,-0.5813043264360428,-0.3440857530049617,-0.2283340049386881,-0.3315015020235156,0.02171875108300562,0.5400744207744624,-0.6427042306173846,-0.2486035705330551,0.03801103339851799,-0.195535048680252,-0.06988740616307465,1.190031699588169,-0.1340185916548137,0.004188476882473101,0.7573435955787851,0.1746290025533683,-0.6164970573607694,-0.3164530479426814,-0.240144429683506,0.6039325151167952,0.4372527379182071,0.04117476802038938,0.2006391302385236,-0.06650455318587434,0.1063845142667018,-0.3224675609385196,-0.1304337525322791,-0.06030468514047674,0.363342690203423,-0.3203777124400359,0.05655097058081834,0.2157829559820886,0.06315049688583309,-0.2775499062950298,-0.3654738193894811,0.5975151156420616,1.477460859395626,0.190955690106879,-0.09253697008356002,0.6495430937684716,0.372917566209341,0.09703328061230089,0.2156579763571063,0.1091436538395922,-0.2614384509642784,-0.3283146457329544,-0.3183666387116139,0.01409976833797403,-0.3215631983488982,0.2078269748916806,0.9252686928192292,-0.1540985068257165,-0.2559503470437809,1.94598326283016,1.499202191183235,0.440117488531854,0.06111508188955451,-0.3442123953184192,-0.03313929811798594,-0.3160693724627838,-0.2199866827744608,0.8610509999333453,-0.1470869486914374,-0.7748641177243208,1.004524945015033,-0.2157610746651724,-0.4441580966230316,0.2988885002414811,-0.1174582998223984,0.02802276671423743,-0.4149306706166966,-0.05011559764052614,0.07952526128034548,-0.1063601458240876,-0.2543725433134248,-0.2754816178635259,-0.8030555901356479,-0.2652330162841831,-0.009483042940263238,-0.1561966413163639,-0.05173519882539172,0.05619624243382206,0.05238432743964035,-1.016069388543469,-0.2710917654712925,-0.9907154407997167,-0.9905113655207284,-1.097541531869108,-0.2675993773977653,0.1844601534036597,-0.2082991629786332,0.6562360372443359,0.9642132782110018,-0.3459398653613464,-0.7085214167901185,-1.250152337028759,0.1015751485387547,-1.49152712032428,-0.2173911181261462,-0.5163204166054366,-1.125480545520948,-0.5651570997980597,0.1866279381530088,-0.3750787769215183,-0.4295665501905626,-1.116269918091191,0.003394179039422844,-0.319764132673363,0.07278032108121103,-0.4233762912148791,-0.3525490115470673,-0.2824488861598231,-0.1362997961943391,-0.3863456826569201,-0.07537166357957371,-0.8900060771957143,0.1550632745404889,0.3470877825707803,-0.5824227555128143,0.2368835965300238,0.8100208670186245,0.06316878783894042,-0.4512397185154806,-0.08091600476050002,0.1643113442954806,0.4508782323373225,-0.3067358556014005,-0.9548171641426515,-0.05631234059994266,2.257719502109412,1.225645301718949,-0.6769610084389478,-0.6797332798853765,-0.5887913495612691,-0.2181550336138505,-0.01704274730385839,-0.4768040140164529,-0.4792841424389211,-0.4446697267796541,-0.1386881448095493,-1.086962756107418,0.006250067334626561,-0.3737298679265371,-0.3270760712832176,-0.600672850405542,-0.1194810389499852,-1.21757142357469,-0.1293647102451657,-1.260694987873975,-0.2079294875462467,-0.286307528208325,-0.3833783028575113,0.4520944703446497,-0.1891621635420616,-0.2833664615406385,0.3446371584540398,-0.2002853760633783,0.1904636665612,-0.8634950641152215,-0.09176686876405107,-0.4597325816621351,0.05439557344601064,0.02486770139408595,-0.07821223783076887,-0.3607825523935487,-0.1881883460551309,-0.9695757140151655,-0.8707386123737823,-0.3684449954527991,0.2854219784876376,-0.321389752753835,-0.6227370576862207,1.5698464512076,-0.1609028693556083,0.3262587529958886,0.3271864584765111,-0.3876945600264384,-0.1857200046719548,-0.3572211671890416,-0.1634697050486936,0.1235714923100221,0.05463906964652102,-0.6366721596385518,0.03208066975385146,-0.1897077908175115,0.9658507585500373,0.7646993458036934,-0.02225671883205839,-0.09684858254020455,-0.655413704795821,0.2268263064651715,-0.172880060096904,-0.2877225749055092,-0.3842777161181181,-0.1422093003450739,-0.4611282121963747,0.2063602931741588,-0.5709761108339643,-0.0939645920810851,-0.5478046810419462,-0.4137126578544238,-0.3418982936508181,-0.4063403454763157,0.1825500608258809,-0.575370955804311,0.05279593118382458,-0.1876746457944819,0.2535963458766787,-0.2430005520534101,1.19659303644435,0.3071301602047835,-0.2884637581951789,-0.228355941371708,-0.05378462307637863,-0.7479337470434236,-0.3769932793366594,0.4084919313815153,1.12709840776781,-0.4087049785579522,-0.06803446249368707,-1.241099841932099,1.30805959469201,0.1008046658297886,-0.1328407578849918,0.04178191475189733,-0.2523270563854448,-0.2777081957574998,-0.1623154391587475,-0.06824560342220679,-1.355718347030723,-0.0106391880744959,-0.9210971607715237,1.268241593673216,-0.1364631852024944,-0.08880224717069604,0.2108995988765552,-0.9878134040965124,-0.5898879193531482,-0.3552125619024946,0.07617340344454715,-1.703247126772933,-0.3430817542439383,-0.3415942131146711,-0.09398384956728892,0.1828249624912665,0.1646478028240707,-0.4589786620363487,-0.6326279803260383,-0.1520115579965368,-0.7330412997734166,-0.2677207966409711,-0.07691282857499783,-0.09686225383873991,-0.2464495461893525,-0.8110012457612182,1.268201511367163,-0.7507584553844576,-0.3602887523908759,0.06832473186153383,-0.3317164570759579,0.740850866753656,-1.425865272859918,-0.8345947969723978,0.1087376445330565,-0.4340799179857696,0.6188004800579922,-0.4247738243320636,-0.04418079642983361,-0.2860330824076123,0.1943337145709598,0.02827580951284022,-0.203217858887768,-0.1739398985210214,-0.1438149135951953,0.5049174249064932,-0.1717163141238358,-0.2411799749955699,-0.05053018415270696,-0.31531752060888,-0.1821361979469319,0.2194279245105482,-0.3734866942727655,-0.04986580892572775,-1.046566568282756,-0.4936105323974703,0.5083368268727596,0.532873988419952,0.09724845112005179,-0.2204213004114629,-0.496071335218968,0.09250037593074892,-0.9399288301585873,-0.6165801068913793,-1.479200430581674,-0.3272990887312553,-0.1796503094814309,-0.4212627931977125,-0.748038795300045,-0.2157875161925054,-0.5751641513929666,0.03383218723669777,-0.2731917541433022,-0.5586187089326015,-0.04231528169583015,-0.2655644801731968,0.2655301073975773,0.4580866599768191,-0.4349605062980842,-0.9350282698697786,0.9199165185036341,-0.3170373118403741,0.8180070754921239,1.657111600001544,-0.4659380225705405,-0.6551675868818517,-0.459625590155617,0.2511868417239025,-0.0589020784459438,-0.3056947382009166,0.153661914606896,-0.180513734449138,-0.5606512194616334,-1.046969529071678,2.232570391793842,-0.7228062949212691,-0.8823412379026949,-0.7587304871275956,-0.8426924243738592,-0.2664154124779688,-0.6308970702975315,-0.5893901189040565,0.04656141754966832,0.4344636905333397,-0.3653565375418375,0.01108993138889234,0.4451563573614093,0.1713675068911735,-0.1156456445355306,-0.3321105260758907,-0.3262325391355178,0.7922487287926757,0.08138146280632573,0.29421178916642,-0.1405856322321459,-0.8609622338620726,1.019306016570814,0.03892572322300818,-0.1960113710690779,0.9390662585298584,-1.00373634711729,-0.9860473234106192,-0.1666927810243808,-1.348210661365183,-0.0426840273229528,-0.238582481806743,-0.3435480948072114,-0.6376529989316458,-0.4179853787932342,0.6996813198178263,-0.3149334171793888,0.7278984864449247,-0.00993443919353218,-0.2449647773147498,-0.2041050422138374,-0.3638653518365449,0.2258941616901702,-0.04414659653157396,-0.1105153455655537,-1.004792858946621,-0.7533142920791905,-0.3180271257623006,-0.547154943694192,-0.1899128623203228,0.3489789392150108,0.455059291384121,-0.3337766987439789,-0.1928222970752191,-0.07654609910717314,-0.3564102321836927,-0.1840073589606638,-0.4643579140054654,0.532490913109966,0.9354860225117635,-0.3296851226163042,-0.3874874307057882,-0.7153496997385551,-0.239730621873258,0.751909753128716,-0.1756538708541395,-0.1561267963262332,-0.5948546349493348,-0.5505541010560973,-0.09191574248085188,-0.1803341044723432,0.1183533073177304,-0.5927894807840067,-0.8579843144268041,-0.513852052140664,0.1206606057782718,-0.303252882495023,0.3154589395201933,-0.7445829900560246,1.221455789030385,0.4878226006792123,-0.7671641855175392,-0.3304318167578337,-0.02543402371692424,-0.2739634271199448,-0.3264037513863635,0.2124165631285231,0.4981660066759347,0.03638252289187465,-0.6070219229126737,-0.1347898942150906,-0.02488929784749135,1.229212220500824,-0.5462785190283824,-1.116545020676607,-1.661448730367781,-0.2132118846706087,-0.4314698141833848,-0.23353510341974,0.08843734083773044,-0.1926267956560216,-0.7756099094926728,-0.02586321134465476,0.284039170619821,-0.266811322422417,-0.3903709584782241,-0.4446836497909662,0.03381414177815302,-0.2099785781041564,-0.3073089268042151,-0.1601727397550479,0.1500403188131808,1.807794840250707,-0.718814538485203,0.03581913371555784,-0.1574271740747177,-0.4130746394288681,-0.1563243601442128,-0.307746646460389,-0.5595308497585678,-0.1918058217183805,-0.1665355221925607,-0.1831904276328301,-0.1996520729546852,-0.4894407229629744,-0.5621634695483397,0.3347982221177671,-0.7984699988297516,-0.2076653189298413,-0.3857807304759205,-0.5529936586701976,1.172091957696205,-0.6055180022848069,0.945621835896107,0.08474928511844688,0.1418225029053161,-0.1555631838639643,0.2130571429545183,-0.4029202520525645,-0.04082427581110484,-0.1035714131034153,-0.8476235566802847,0.1143607996540904,-0.1880837267802101,-0.8795471009914112,0.1897090879863437,-0.2857465659117294,-0.9459999600469722,0.07621055457615164,-1.024964126775721,-0.2947898078552567,-0.4058167675629678,-0.6205920702559456,-0.5518601278692511,0.7675494429868878,-0.003347333961277218,0.408086831892465,-0.2794788994995104,-0.3097929831415699,-0.2447525449843615,0.5971685522188303,-0.5259542956693076,-0.3847113860640245,-0.3607737854059058,-0.5553787413326098,-1.270256670031803,-1.3268010742806,-0.5704680782081974,-0.4907866085378885,0.04111289439883675,-0.6713696598207368,0.176423650606612,-0.08968072188034883,-0.5872573535386709,-0.3690576712334219,0.008926130722893789,-0.501098058774011,1.883124738090652,-0.05190992288454404,0.9366553524912175,-1.470137303767144,-0.0922886832408651,-0.8483884054447585,0.2802289088837016,1.319791193913838,-0.6059888842517401,-0.7984940938432157,-0.2425111996910488,0.665946728931094,0.09092852469367724,-1.010636952807183,0.07599255166715978,-0.329040110570988,-0.5600408501172444,-0.8190693828766269,0.496365177740112,-0.493403932832025,-0.7712876635494226,0.3018021438559886,-0.2848877564303781,-1.325267441390444,-0.492161409616171,-0.3140987859665103,-0.2055326085400943,-0.1158952678688527,-1.956694581901011,0.683120868972123,-0.6466247495983896,0.4148517178835374,-0.3019178974912946,0.1672803053014437,0.4640811806010163,-0.3218095001087671,0.4241985535820411,-0.003337392728228735,-1.210750546079308,0.3000110740334238,0.818521224829789,-0.8559440680825934,0.7046139508288736,-0.6589645689516642,0.6507367097743388,-0.1817990688929202,-0.3107298410043182,0.2833018605802309,0.8959745228945273,-1.432388603375085,-0.4726630190987763,0.06882627630658951,-0.1241133597304279,0.151225548795449,-0.5071116302544353,-0.2139389720940866,0.03242600190157705,-0.2676790903514768,-0.496090582855001,0.5380532072301067,-0.303111997633722,-1.394536879568181,0.4519511455905802,0.5717173991563202,-0.5575415215300956,-0.05186401380231923,0.1740184139541238,-0.06498438774927119,0.04950477543319348,-0.4996925903712799,-0.2260144526577659,0.3553177483312592,0.01750265173996847,-0.01647945582441347,-0.3110692786660493,-0.6115507293354872,-0.7738691316265848,0.3038840746825872,-0.2024340053248392,-0.6371469759157438,-1.325475219587288,0.7810307994095321,0.504077003855412,0.3332716544509109,-0.4266127265302482,-0.663273396176007,0.9374163688099472,-0.6883286678488238,-0.5512108316303599,0.2906310009075277,-0.3952582018811181,-0.5242916215568149,-1.266561435489691,-0.04494400453326713,-0.3669341538218548,-0.4942061671561388,0.6771100909385996,-0.2152057395339002,-0.1332913386781419,-0.260061071107986,0.05028757212410704,1.819888109042565,-1.043226470748751,0.08393088419416458,-0.5785951860624393,-0.04295156423032175,-0.4983045639310593,-0.5207739258858743,-0.4123435802651225,-0.4742343451490658,-0.2248041952979868,0.08290135862588786,-0.7871980194984708,-0.7656398352312509,-1.009008891710971,-0.1912163148614207,0.4573849212250916,-0.3663349569074602,-0.6330566755376577,-0.1438007046711,-0.3491792028874108,-0.8723440061257967,-0.05638781478021868,-1.535580529916206,-1.148982550282755,-0.3622736769337559,0.7148751197164502,0.03747786345768772,-0.1301962156936539,0.4686760981689452,-0.2624432029415514,-0.06657284694896713,-0.2903358623529199,-0.027693766136116,-1.07167963731235,-0.6366231604611936,-0.5548857943494562,0.5840760661298645,0.0328585573081042,-0.6488599215450258,-0.44049631609078,-0.5703430945330306,-0.1294412905474936,0.01845523991137739,-0.616158167567724,-0.5386124660770842,-0.8268753462633976,-1.156749397671889,-0.03441580609642102,-0.07033144168942564,0.7104838571187432,-0.3658050949227791,-0.6348242052524523,0.1662476917307817,0.05601987476027863,0.02755820938191706,1.419485360188348,-1.080077027732788,-0.5389456228595014,0.9354551693169914,0.268157391189032,0.2560739685951424,-0.4040874443003914,-0.6290693320492813,0.8905365164027658,-0.7435579788307954,-0.6426274306725753,-0.6290974187936197,0.0220241358396719,-0.5662578100677078,-0.8704087382701211,0.3662557611056257,-0.6600637742027896,0.4735273609497688,-0.7530053340722646,-0.2617753620696929,-0.3424644668641514,-0.1297049015953959,0.2683761869280781,-0.06419128907044593,-0.3600401413428098,1.286568688221566,0.1011009104322545,-0.06010114010298422,0.5739718573351051,-1.101549561326225,-0.4954036452692797,0.002583212279947792,2.618259563674225,-0.007917811113094165,-0.6065179152735976,0.2821646825224933,0.110042956843992,1.548428529884932,0.1927429747652884,-0.5039375346359349,-0.3074222837394979,-0.02413515063716519,0.4672922800208108,1.276245239133911,-0.02292797800783734,1.392381714251645,0.500281296173114,1.366026792981413,-0.5016193377438279,-0.1580164931127079,-1.01880686793857,-0.5220021745040474,-0.235396585898186,-0.5589483915342789,-0.1626143345198122,-0.8161256909299195,-0.5945549935037867,-0.1523544505386787,-0.4986274291704894,-0.362393200612548,0.6261535419580272,0.2453036130321373,0.1009272801977431,0.1165573340039662,-0.5068805630121619,-0.1828918904326187,-0.7527239086618144,-1.305030680346778,0.09617978175441724,-0.6211686590976799,-0.2769014909649253,-0.044002874648634,0.2281638527182061,0.06960991620304519,-0.1711921314841185,-0.2499043778701631,-0.3708451967078408,0.08038755821901659,-0.3016352687987023,-1.661403990921091,-0.5256233543042382,0.09276404381474669,-0.7945703420447683,-0.4271261383337333,0.09600220592777181,0.1546963875364946,-0.4644423707146867,-0.3446900496840737,-0.3799866333104102,-0.5060738004247437,-0.7834307004580493,-0.2249924841098238,0.687539045450441,-0.4383464288119706,0.311867277390861,-0.2570331411611909,-0.01592641011312319,-0.1487776669286325,0.3531572068340632,0.3458346309166597,-0.06033452364295523,-0.1051139563338096,0.03236468986362466,0.3525326854494312,0.3530325837757119,-0.443742869854381,-0.03139941884490328,-0.03520983842611771,0.01659308537160878,0.4648605373629141,0.4006934083283645,-0.3859011556945669,-0.2863285333917603,0.9595567065786681,0.1031149403684539,-0.0003598492955016424,0.4938093617010793,0.8583413103093197,-0.5123309940404023,-1.27866177118606,-0.1789954722640188,-0.09060789608117906,-0.4818877886734728,0.07431613361519614,0.5817186741837204,0.04934965494236796,-0.7365438180498505,-1.627967905558845,-0.3299982879307473,-0.125805921967982,-1.524808729956005,-0.2871988848081169,-1.634387652004743,-0.07709087435106177,0.2282926768117693,-0.4896356461751235,0.9687176093356545,0.2391595078890941,0.321886541914376,-0.541303883457121,-0.03860727157178755,-0.1219975475121193,-0.7931077307261015,0.6424595622050459,0.6810992636124876,-0.05577576993190091,-0.8789924302743487,-0.3163310649549665,-0.09303760118176924,-0.7652069055623694,0.005904677190533136,-0.3079615026437277,0.01541974456170437,-0.1585815456877079,-0.1526102473588589,0.8337664970844824,-0.31696606091811,-0.7765660999147977,-0.01181870393335056,0.1717097996566086,0.7524573922064378,-0.3586467478202915,-0.2446614955770192,-0.5133839666798753,-0.6161501339008822,-0.9552521816804607,0.48985206438927,-0.4702567181882502,-0.1465590791621137,-0.0339531078507842,-0.732843206902323,-0.2778892338076734,0.2667313119761207,-0.4843798336740556,-1.043204736939011,-0.4464439242796018,-0.4239612551051701,-0.8325465043473583,-0.4392847705217679,-0.9896602994104461,-0.1273391531500569,-0.6053005225227847,0.151671621315142,-0.5494860983269136,-0.5734380381223367,-0.9760839904094157,-0.5413306935354331,0.07430523275215528,-0.1709003131638143,0.451887120613464,-0.280921887293701,0.00442516513353719,-0.6513650620116983,0.2281407426690453,-0.6114609172526966,0.04067211973073951,1.579426361779305,-0.3346440412901376,-0.5059174034059039,-0.3087957648037779,-0.2359032777779571,-0.1425555714514336,0.03561001970925861,-0.1658858980057448,-0.2162544965945969,-0.1503718735563672,0.0463256158210916,0.1833199776463562,0.00641346921314363,-0.3302705693548491,0.7144450658562799,-0.2901464314887575,-0.7968038465675228,-0.7689546762667969,0.4690742007146056,-0.5557819966964764,0.8756178242305219,-0.8205110107996235,-0.5112630331440015,0.1691027388866114,-0.3603595126032054,-0.2556528284540344,-0.2806662009670858,0.2328099965930804,-0.8442902986143731,0.3886151806824594,-0.6090235506051235,0.07691526487977449,-0.3392304865021013,-0.09058629552989121,-0.5411549318617201,-0.5057747160630162,-0.2748858234850992,0.5477066159504642,-0.2841301763315884,0.4363563683955893,0.4408041041426229,-0.3315204824570631,-0.3848614316677658,0.6916624842770612,-0.2367957492609114,0.3192921853603245,0.4129805795474548,-0.01137463969446948,-0.2787856382925001,-0.3040327118473131,-2.087211445005932,-0.2519416333805684,-0.5408995106008824,-0.2151820331102985,-1.02420609328956,-0.04559963048368485,0.2614479786469718,0.01257208047069933,0.1311801338803715,-0.2236324338932869,0.1368236660437836,0.3547035835552941,-0.8358551649897586,0.2355351686752479,-0.7367891904299274,0.3286915867626879,-0.1520497082305979,-0.5321991232375642,-0.9255378092586972,0.5313276559520569,-1.000439868961544,0.3619613719200093,-0.5102874589353583,-0.2505325837294706,0.4314703100230062,0.4227866994215009,0.6142118352383571,-1.505866118525048,-0.5827599289566009,-0.3671304833675712,0.3058552184569432,0.592044551622959,-0.2359171095417011,-0.2820404849635272,-2.200168425514672,0.404095863527569,0.3867696211589529,0.4345357016998579,-0.2195447877987285,-0.4523235059768312,-0.2277331659663669,-0.614565246096307,0.1955513116646526,-0.3064089687313857,0.4201752699508725,-1.238916051992398,-1.049463910049883,-0.1492315667586616,-0.03656156458798265,-0.0721093527460227,0.2318292554123287,2.529746645380468,0.04731248489926741,-0.1323264475519443,0.08091474009407809,-0.2179223621055494,-0.5113120069936669,-0.7596596757271683,0.3661531668909568,-0.5076181255736002,-0.1542678777609146,0.04923946185348999,-0.01869488949106791,-0.1319899533680922,0.5945081341007797,0.02981884444183319,-0.03337516602631614,-0.7964661427200561,0.1882642525962109,0.7422445467524693,-0.09256388132460031,-0.9449524340961217,-1.206109301150552,0.5580887659890899,0.4532820042134232,0.1742294897793846,-0.4260271237316345,1.459297513391082,0.06585877174265763,0.3391419430512992,0.3730963685334049,-1.085054375337336,0.02354218894424413,0.423415605812324,-0.1801858888176825,0.3364705369400124,0.1266719863516101,-0.940555981815242,0.2553147098716249,0.3222120481290771,-0.9508825716707178,0.1541211174659484,-0.4310377860331067,-0.2314947893137025,-1.214855599832155,-0.4030926586661269,0.550290168454887,-0.1851678124853887,0.1386426708667083,0.3896400537540937,0.007569003738686473,-0.4526180556782351,-0.4964010302328202,1.200934487183818,0.1578525211098719,0.4952507323022065,-1.153951294700945,-1.01803960651774,0.4665919139561647,-0.01161729698374048,-0.7951502193124773,-0.3509308650533459,-0.756639900754539,-0.8426964965212846,0.2341872789499826,0.3115361897052717,1.576730962255206,-0.2522364212299319,1.756562242184133,-0.1674282439051377,0.7111872172355117,-0.2198468354062198,-0.5038976060070339,0.06731912626450488,0.2750779781888605,2.183780893902264,-0.4489394918915888,0.2483674619973059,-0.1189575339187775,0.3469784288382823,0.9015350997751886,0.9410896846955907,0.2275408117510712,-0.6555352956807218,-0.3788037213232426,0.04881864839788638,-0.2015067802623255,-0.3922364238123817,-0.08557649268417306,1.736001817845217,-0.6755730632349274,-0.2286885093098906,-0.3421319270691541,-0.8805911205781233,0.7361036318438999,0.4673935643657236,-0.5926926409502182,0.979650014043839,0.6037634360440752,0.8555472930639273,-0.3681990855283722,0.07711975202359742,-0.4882013980936071,-0.1414091293677406,0.8883331079800461,0.104164571557398,0.5164029983474786,2.732290884825074,0.2663041222812857,0.4297013717212232,1.330935462740118,-0.2466485064629511,-0.2330661986130189,-0.0805526847971236,-0.5452814563022383,-0.1711492056058315,-0.7464849159509045,-0.9806912016348259,0.2832431909214947,0.9073038461123993,0.08893445358420854,-0.8157797751290158,0.7492559151680519,0.5051516846070198,0.07849715015787737,-0.218481460134511,-0.4302050604983941,-0.12273378883054,-0.302683311989579,-0.6123494066553181,0.9132988054006701,0.06752505373321595,2.279201491103438,2.071196159207491,-0.3218374443376436,1.349736604477605,-0.3930715518880266,0.6031553803262287,0.0160084396436339,0.8311047881068282,-0.3159965641738494,-0.2171577998417348,1.702536597354087,-0.6187326781339147,1.701094429500603,-0.8221181305776825,0.1151471639409806,0.1073454543134022,1.638355651231804,1.577258679065867,-0.3162727919093096,0.4236050991772116,0.5828481225663138,0.287530052188311,0.3029573944243896,-0.09172040884955715,-0.1813739405068684,-0.08387096966806665,1.041819714174734,-0.3843926565030171,-1.048745636295248,-0.5360331539545771,0.1681172057522548,0.9589086838459966,-0.4971318324652806,-0.4948259546486347,-0.9261050103440756,-0.4725468718094347,2.88814225604399,-1.005539518420687,-1.015548292263612,-0.0533277966655414,1.819366568506056,-0.04907820821282029,-1.110503559172545,-0.1465357001416232,0.1259869307384095,-0.1975077346335184,0.1082436199691732,-0.06265006160414108,-0.5312147750973617,0.81132853148091,-0.1307127456142798,-0.1919553761573146,-0.1114565821255947,-1.086131081180299,1.349610964728373,0.1165061390649978,-0.5749545649419491,-0.03052726042362694,-0.4703986227634427,1.160315607026627,0.07319228411027373,-0.09406262995433516,0.6361967538370957,-0.124729015157492,-0.100901978843375,0.7015658641962715,-0.1569333664986537,1.272391488449376,-0.5533612754530429,-0.8635159080940651,-0.2981329910614223,0.03507479933361632,-0.05711166208080337,-0.2553987155202427,-0.3744649286475394,0.06585877174265763,-0.2604818660542991,-0.3404784721849511,-0.2127524737601706,-0.07440027157643918,0.5583924942605852,-0.407639714500435,-0.05758919635391922,0.1261150364607108,-0.4353897649162158,-0.3063986558985469,0.2171990560047027,-0.7782168316116544,0.705960021126944,-0.5460606671751287,-0.530294136152542,0.1187069763174463,-0.547477848825471,-0.262449424915446,0.8885417979404496,-0.5667731252725297,0.5008269500097098,-0.1724064689142861,-0.6956284156260388,0.04290539467318441,-0.3656713149729068,-0.8637234062938076,-0.1347098499121232,-0.5035153964650996,0.2504744975926045,-0.5933806383313343,-0.5848985020779739,-0.6891825622512541,-0.6333074445279935,0.08108616936484889,-0.1633263806133752,-0.03615049163162785,-0.1823526659180875,0.08031037927484248,0.0733698595957718,0.06005085210947363,-1.171615957418941,0.2642580515028667,-0.03002560862658362,0.3152098410062206,0.5136035083709317,0.01434603911010664,-0.7182615098347893,-0.1819179168480516,-0.1480684929784238,0.6561005488109503,-0.2416501344369719,0.9988538974399583,0.3388902094531709,-1.019853193680053,0.1665825361551612,0.1576134450781228,-0.4333847526321939,-0.0878739779772163,-0.1918234635441676,0.04499166026517989,-0.4351796384868359,0.8017771815737547,-0.4255701697067305,-0.0704286445868907,0.06373439580080166,-0.1439192754863985,0.03312908282327815,-0.8044186143233376,-0.2785638535042653,-0.8429695138292169,-0.1429233674700537,-0.9194627076616848,0.6528699832962515,-0.7811024635769687,-0.3291417217792523,-0.6729197472402425,-0.9360358788775076,0.2237074901104537,-1.531000340829803,-0.009067817530214656,0.06043570685784042,0.5850970787646614,-0.01705966485102264,0.1373313179994164,-0.4698728328516322,-0.4298757178457392,-0.2385255461384116,-0.1271113508597744,0.2206697394312955,0.2909009240927885,-0.1743765863236096,-0.519509314810234,0.79947773889123,0.7121541962424,0.2705748982153682,0.2603807445699641,-0.2107417218822907,0.3076919031612578,-0.6100644273522094,-0.4434009080119642,-0.1423898669958897,-0.2349447840650313,1.381748262905187,0.6873772450562644,-0.2230376470326647,-0.3456934947201964,-0.2424462885920018,0.01274709666329634,-0.1674391506344829,-0.5935946185348651,-0.3998437967698936,0.438011658095636,-0.04374050903750591,-0.3500501233834343,-0.4310282445027831,-0.7852667428849525,0.4167779430358929,-0.01566319950137571,-0.6923188917248326,0.1076225838645484,0.3207426791059272,0.3135193150476961,0.2710192424892062,0.05151496001342345,0.3515863786194264,0.6602149988715535,0.05510561463236843,-0.8256190422957553,-0.8250867764654511,0.1351408807943213,-0.6190724592032723,-0.6832790015925718,-0.5025730513226719,0.1060890837238866,-0.2643101397014666,0.6489680338430802,0.09021376752897448,-0.5451313125238094,0.05588675474250353,0.221594733023382,-0.2997967879223016,-0.5075203639654009,0.02944986461615821,-0.1213625846921876,0.4990332798158514,-1.031887693502466,0.2471957204532706,0.4447683603020133,-0.756639900754539,1.065328096139264,-0.1807596841774217,0.9762491142770056,1.503617691609692,-0.1478416199356344,1.805273755050961,0.3004936549206943,0.1421829514898134,0.433155835162973,-1.569890391913437,-1.116916263335869,-0.190820448442851,-0.04943843086140285],[0.0004420732510630734,0.0005606863663858236,0.1451077924354758,0.3252258017280888,0.007743352627256711,0.002092748993179149,0.08659882832788338,9.575898572679851e-05,0.1534092123972021,0.0002384020182991507,0.1553617185225007,5.616479144108348e-06,9.680549188474556e-05,0.02552059959595477,0.9470223838816275,0.9363001901663514,0.002098992236046552,0.01083166000522378,0.3844348037935529,0.004104633744265633,6.690813851321631e-22,0.7104326441437273,0.7024750835677513,0.2872203394771029,0.5671854348302074,0.8395577584309113,0.003810773744319425,0.00153265630655527,4.974432399658044e-05,0.0004608364480513965,3.459955466002547e-05,0.2448627722712622,0.1184898960339169,0.002494187636728363,2.331843035756075e-06,2.199276230849002e-06,0.7756918544557035,0.9553308424446226,0.7624007791915542,0.0153958643557085,2.192915974865687e-08,0.1942477317118765,0.005379047753630566,0.3132671256265559,6.93661803111618e-06,0.702476780647551,1.99056429856091e-05,0.06671261679566803,0.01787346396478454,0.3866202269640096,0.9950683017310559,0.2238162662523828,0.7456409759492744,0.07575346427819955,0.001634160939335536,7.022366209741949e-05,0.003282180458673444,3.085210714573983e-05,0.144759153299908,9.462567284812834e-07,0.02584367448124049,0.01063474216294485,0.1408344636840606,0.2194285786332573,0.1272003745289423,0.049994196449693,0.6804202073420909,0.7522975374131476,0.3360553601094302,0.02325037328663737,0.0001813148325902249,0.06420136520171831,0.341964106776812,0.3554216469151183,0.1733973733718484,0.003152157544588632,0.3488614770677054,0.5009667981935684,0.965386520611962,0.4963401364887789,0.3283478648176285,0.007500666419494252,0.04694392939284727,0.1000298811927505,0.7737022060385916,0.4302386137689566,0.0001853110641323305,0.1529485645208909,8.526973361509948e-06,0.005729033617106441,0.005829897841558014,0.3751758161675854,0.003204862271581751,0.01918989946153765,0.685339599623771,0.6347466478846551,0.0008219706540293736,0.0005176568794042938,0.01594761461364107,0.02919079977563745,0.7254024994802248,0.01989275306513128,0.7161072023478816,0.891131492131317,0.004435959486785619,0.5205568490135665,0.3946826669058223,0.4713483926263014,0.003311761025680209,0.5741041593356259,0.6796586881704092,0.4679532200307828,0.08678939257864469,0.5049080943446438,0.08659059589843288,0.03221337099609316,0.0846836310709473,0.7469992571282166,0.6290888415512228,0.04310241033653471,0.2675840413803131,0.01540039957783161,0.01209237074023552,0.09896386267069514,0.3884673228481954,0.115572068920251,4.435567715933691e-06,0.9841074692881232,0.817519403467471,0.05302009812947486,0.0001174951431580521,0.01836071080471603,0.9326677445173869,0.6904074589788963,0.8429640734532976,0.50346629193641,0.4158128845921724,0.6018786272660255,0.2464958579027009,0.3123379061971268,0.008248501045089665,0.4876646992206556,0.4176899663973457,1.051359229717726e-05,1.917192729894771e-05,0.0009373645207619825,0.5135484133306357,0.03869862189813477,0.2958238119667142,0.0004980780464831053,0.2194453904220377,0.02020978875544378,0.1566237553056567,0.7898557883297671,0.001021692297572724,0.3611652623213838,0.118401338908366,0.02199195275006046,0.006019424751590049,0.09746526676294028,0.003969673229446694,0.3233825711933948,0.8570243305259124,0.02057856261065683,0.6074317507438055,0.7277447962942727,0.1773473096763716,0.9875414512444294,0.0004973564511475256,0.2059345667913449,0.03691842186833229,0.2041713892987258,0.8483309320977116,0.9689725960404636,0.0006479329590245059,0.005416000771285095,0.1074831297125436,1.735117476480303e-07,0.3566051426368453,0.4758547441119819,0.2494621148657269,0.5035777996887982,0.5589062070817157,8.021089946030685e-07,9.568958047832265e-05,0.8682273278489838,0.09876223338805783,3.467717556082736e-06,0.005730051497529126,0.2869960541453584,0.4001557657242756,0.002113632427767676,0.6253876628777095,0.142512111841996,0.3107547020609976,0.01505441814666458,0.2711580328389318,0.8137420783296719,4.627773639752732e-05,0.002385347154295384,0.121839339157427,1.061275681979225e-07,0.001168304961997119,0.3492514625162512,0.03606217933551654,0.4562599421848899,0.8257253795003558,0.06818026995287874,0.02015593279468529,0.08778162626067634,0.4455226039416476,0.3679139595451683,0.1483573306207634,0.004317360764355581,0.4746270881685558,0.01733803123082684,0.6649787284657631,0.3482160944433001,3.306430373842624e-06,0.8188129905540558,0.9800128896764549,0.6238324278270733,0.7463392415792983,5.223221147040451e-14,0.4165884557132406,0.04693533831831481,0.1251755709966701,0.05723838397106413,0.05385687490705184,0.01309901611115658,0.0001232211217778975,0.09966235153942408,0.6503831033907601,0.0002340158976404387,0.009197523927924292,0.0002222613216084426,0.03411276925477202,2.557705318729575e-07,0.0003444929307388046,0.05306139074232472,0.0001166313683451042,0.02755976767884849,0.0001068310674208404,2.873495019637346e-08,0.06031841453640261,0.3024415450699297,0.175084877969958,1.513156813642286e-08,0.001952330461271506,0.0634861460367923,0.06255954945607742,0.009972158136624621,0.7538192447436792,0.3529513903801547,0.1079258649907685,0.01971132750007707,0.263569416583983,0.015177885216423,0.9355401321779246,2.143551184580172e-06,0.1048420261583337,0.1978983495348146,4.844928066054742e-05,0.0590204762506843,0.03090645522550706,0.8786547476026786,0.702476780647551,0.9683282131442523,0.2009338607468115,0.01341906218687159,0.2966258022542025,0.5810604801569503,0.7203530045103602,0.01769102253178155,0.6965228948880448,0.8587222667568554,0.001271173478540128,0.02165071717802354,7.513093251547607e-05,0.8692619594240978,0.09688090157430468,0.8475762831286417,0.8360469908713895,3.435566432986046e-05,0.008420035563340696,0.2933884825668035,0.3026535665055229,0.9213135860994142,0.8580581648319041,0.284875369091671,0.3474869603650558,0.01848421914371141,0.7813552650888332,0.0367195988357507,0.9745352866381047,0.3887366352748619,0.6588260599063943,0.8682644988610602,0.6779049889984184,0.7808585198434491,0.06266036604048868,0.4302386137689566,0.002614074672315393,9.80947901133322e-12,0.1318725787806753,0.006496352114273727,0.1489173671140057,0.3385436847582683,0.2080727240190052,0.02604826927623857,0.07845346103704536,0.4163696585370026,0.2092920553697732,0.1986421645848077,0.1079258649907685,0.3376204676051383,0.9411477802288701,1.364427735721428e-06,0.3011209215210867,0.05341621704439789,0.2675953985814183,0.01364333474159973,0.006283994460024314,0.6955119184747615,0.06797683547592892,0.09884116790192599,0.8016178508002819,8.8802083362042e-05,0.3157161739875554,0.1725796953569214,0.06380994482759116,0.04592044264058315,0.01130337176836267,0.2536755400053454,0.06916573383298519,0.3488614770677054,0.3620820569131004,0.008594274021504516,0.3046655010094583,0.0001954632326032048,0.8261903806643506,0.6123757152222223,6.237988812512002e-05,0.00842517124145503,0.00790111300002457,0.00333212404188128,0.7556201944954749,5.44717670391561e-05,0.3599485781567357,0.007861697038926918,0.2847556939430175,0.01353728121198312,0.1791136343431243,0.7650025613265345,0.3116321976085482,0.3271575629329581,0.6577509541728099,0.0002969510335642436,8.257722617641057e-05,0.709080734269152,0.9028178605484292,0.175084877969958,0.9437143657736832,0.3778609051326162,0.0005897868357079585,0.00161149899199864,7.739017934884888e-09,0.01051241229123954,2.873495019637346e-08,0.002664675851450305,0.3041628398841451,0.3926326153639165,0.563654875639284,0.08955579541609134,0.406951017706053,0.1251079685556788,0.4459447529764113,0.2458799813859602,0.06428211804322413,0.7399918847638178,0.00665828734010428,0.8218276621854582,0.6239418654271774,0.02113558719364333,0.5425125828099764,7.40534677135557e-05,0.9107085811692675,0.05622035307376493,0.004209872030890247,0.001128331855773221,0.782827271638073,9.053858887843492e-12,0.002269770729408341,0.06618578361263845,0.3789512860805644,0.002997861724723028,0.001983226733929673,0.6734465217287483,0.07753011556843911,4.553928851143984e-06,0.0002007171621704058,0.9344598995776227,0.9724916132803773,0.7839799108285157,0.0005972873349237474,0.8831071046183936,4.147749749120688e-05,0.6369830690181673,1.945846808540663e-05,1.735117476480303e-07,0.04590970995025119,0.0001127757369934149,0.07777627014939588,0.1995972718681258,7.088808865754061e-07,0.08781228664517737,3.142567743079015e-05,0.1919631176380787,0.05719921469917617,0.5141359117570087,0.6167427945919683,2.429757616913294e-20,6.990478868004981e-07,1.726691461428792e-07,0.009447216629017352,0.9759502399693645,0.2447286692703358,3.824435514307908e-08,0.2974117790926262,0.4880311325945096,0.5119125454129456,0.3074974770720146,0.2353418382068505,0.216342159182775,4.630205063199288e-06,0.485200447378414,0.02860163106814795,0.1402589844030567,0.9968794885116413,0.2625583135551573,0.6128678725729048,0.005169894894808066,0.8197058301033154,0.0003213889032017145,0.5027618446430968,0.0329664968328332,3.831816594161532e-07,0.3648885995861608,4.435567715933691e-06,0.7641975331731772,0.1835998630179677,0.8589355940303313,0.6114739291547783,0.2522821225712149,0.1692749099649995,1.654201985414966e-06,0.01889804308835782,0.8601333672446966,0.4195573989658394,0.6990432563211367,0.8904781998823377,0.4321439232705952,0.01968681564600242,0.2624623475637744,0.3264513171104193,0.2666749063864404,0.2285416371260276,0.002833068046457967,0.5139746536968679,5.213704456081419e-07,0.0001708226272146216,0.7699449823989331,0.0343343799959688,0.02358665621814221,0.6525106319864578,1.044997347094915e-06,0.1002473659116109,0.5875731534680104,0.7952015460966972,0.1604460456066796,0.1128974612775209,7.583725475715923e-05,0.616377488731725,0.003048475033847762,0.2825034703203391,0.9455345234220723,0.4420509286511208,0.3799449290884507,0.1422028389483484,0.6618062419101925,0.3385351087158776,4.531347385163672e-05,0.6278371808528178,0.1254899030873087,0.9107085811692675,0.7307474606019739,0.6512341572476731,0.4646134306246588,0.5398851320549257,0.8827579323579503,0.005378990934961348,0.005052669116114772,0.0007898864502412923,0.2664980271643396,0.4341785413633041,0.0173114035389033,0.3685678458001914,0.8696042275903381,0.0001344860147163817,0.9143856868525048,0.0004177404994757411,0.002816319006144605,0.3113002272331163,5.784079348932456e-06,0.03255810322949605,0.1832453739285738,8.8802083362042e-05,0.6588260599063943,0.70558544842823,0.1512035493147096,0.9801915768125048,0.3097089317728351,0.7305521761820414,0.3447587070618731,0.01461127570336541,0.04082156675294409,0.2744573366893603,0.3294198849583681,0.03800882807219412,0.08023505938210779,0.6562190947335926,0.03247962570785792,0.7488768635144775,0.02406687836560074,0.05727784788014423,0.5049214732390837,0.9391942456902709,0.0006209057978954071,0.8366011540912507,0.1797697610722665,1.733407080802705e-05,0.8183346069199274,9.252382203324682e-05,0.9830083355982774,0.2633279341769861,0.4706698355840929,0.07601651165556553,0.04589397161357663,0.00543426655003727,2.406343763566027e-05,0.0001614602011497689,0.08949166879167136,0.1243968445682161,0.7087310203966414,0.8900179546033162,0.2476878381786304,0.009569206768556928,0.004057909245011869,0.0001474287041595463,0.04254379875949836,7.591339120497679e-07,0.08289829887365267,0.5263919592273507,0.8095267683749522,6.835110128190038e-05,0.07442916331788703,0.09427304838261483,0.04035963661196043,2.875838561342998e-06,0.3748891223228741,0.9039735535263234,0.002776380813577437,0.005016785604404286,0.0144506273449295,0.8804273093481814,0.7276449219796548,0.2649373732600708,0.6017079203521837,0.2461489340887902,0.03829137338303805,0.05680414128839762,0.005768141293439237,0.5634842775402725,0.1106476640627402,0.8921207002874609,0.3017782884105723,0.9477572293433051,0.2626737613994291,0.6940732534165956,0.7732083577302484,0.003071652810115751,0.02215469730855997,0.07332081959044491,0.4103223072872096,0.03388061490253913,0.5460568103294948,0.480134553061943,0.3639993399288131,0.06690424062639191,0.002609449736236366,0.4673421874440409,0.7711173374168176,0.8389126253324749,0.003663896995773937,0.04592044264058315,0.2973920882586489,0.3789512860805644,0.6855893895396318,4.619530724634755e-05,0.257042901238592,0.9724916132803773,0.08907739503722087,0.0172954448819027,0.1032304393430145,0.0176424568000564,0.6436442547344706,0.04881703013032549,4.029330936089426e-08,0.118761966452148,0.3616531057461598,0.08036340945125769,0.5438413411476832,0.3275456380420115,0.02963973094658087,0.07591823910910336,9.70009401314912e-06,0.3969195157599367,1.552889955045473e-24,0.001490859994475818,0.7319360612239163,0.000941205569304234,0.2430410808951984,0.1725648196497147,0.1803656306191921,0.5004691399282218,0.5860939943204144,0.9280306179056507,0.006959230360554255,0.006698584249898688,0.489683269998666,0.02681932226667449,2.744197682454452e-09,0.9261881631239571,0.1720191477090545,0.2292160063115338,2.040789048618624e-10,0.01025412018773673,0.9133007589475304,0.6178898645084084,0.00319063185836637,0.007843253503907589,0.1687042396126664,0.2539394051212948,0.9430506919012274,0.7325875379566795,0.04600746943150906,0.03907329171209965,0.3667337262441406,0.00317739071482914,0.5905007907756435,0.5837594522824288,0.2442690119514521,0.004974337261487276,0.04352263087833079,0.0001550647522307484,0.0002175516074657788,0.338656482729815,0.0937782208367353,0.0001412383694349414,0.9964906732910624,0.5894951975520228,0.602620763071661,0.5175629388112672,0.232325613835414,2.184813969716049e-06,0.8118030840288395,0.104106307496698,0.2690906080931481,0.8981768256616506,0.03983642558904834,0.2044307722306945,0.9952594379297782,0.9680025018692554,0.04385438664719819,1.923761096322634e-05,3.675286997564207e-19,0.9323579924296056,0.1360629914994124,0.5951546361533844,1.168078854355411e-05,0.03841426886150399,0.2906833529567041,0.2027044997489467,0.01607962422725943,0.2429304533282035,0.1912230330597251,6.210945021295179e-11,0.7865512761003964,0.08562725925221013,0.09804818143215575,0.001652464912006644,0.004032525856076418,0.9996753607380219,0.6348596421810204,1.181759319702829e-05,0.5838104091168859,7.54035708817951e-07,0.5880107125863565,0.6589280056993896,0.1404365762234495,2.693820991083196e-06,0.980730398142588,0.04722652735245454,0.001656980345380341,0.9421117100691661,0.07645339536837277,0.2145007300877602,0.2952457698565443,0.0006783402144011476,3.237864997719074e-06,0.01052604912373962,0.1644725329877531,0.8466977292386075,0.5419393293143417,0.3550328406975279,0.008014402286258387,0.8992379742616347,0.09749008764300764,0.6585863566467458,0.4263216968645138,4.740906373601423e-25,0.09971699151894915,0.001179746516032765,0.08781323109846138,0.04421026831297083,0.02060554055956849,0.4268853242862341,0.4679532735307037,0.03948985300503204,0.2840849407470517,0.07285792826835158,0.229952590463935,0.3683584442140798,0.4813998671624879,3.172571061526912e-06,0.003674766961160839,0.1329660461962601,0.02860163106814795,0.5022601725942661,0.1051383107102665,0.0006244274714193767,9.473022538419601e-06,0.001992023329794099,0.2695026300885861,0.9197626857534269,0.008619349060766144,0.0557262019983849,0.7239679709107324,0.0003805055230330722,0.01889804308835782,0.4973964033983564,0.8736766239628974,5.843493145520748e-06,0.420178485733589,0.1011761090346731,0.0008645605720621516,0.7625793502589814,0.02822192943014828,0.03047566517153792,0.05427222342962008,0.04031563274552735,0.0003424504202112631,0.6198146201822144,0.01405175520742722,0.2777150658560265,0.4595775474133016,0.3549299340497003,2.009973759195511e-05,0.3201568560559238,0.8528618293882868,0.8456978859331371,0.1030330091477769,0.001248300312678912,0.001297044945077454,0.2871819723260652,0.6229970609076578,0.7570514425346156,0.4207469124371947,0.5742413023572805,0.1168865565817889,0.612455622486923,0.07369612671321857,0.1427408831848306,0.01918543313870086,0.4453889984799249,0.154032606192846,0.7722388421342522,2.080868042112423e-05,0.05431857380181345,0.4742622567827278,0.02736461088690235,0.002322936629200693,0.0009808729113178767,0.03481077305827066,9.154044699492344e-05,0.6353306489224246,0.04550330888203865,0.5887725127913946,0.6447659665917317,0.1031249298997788,0.2438122303386971,0.0650503220775403,0.0001122491436713429,0.07562966388765413,0.0001032579626659696,0.04115230459738657,0.01099845351792439,0.003855374507534345,0.4821772091282238,0.0002492267663361914,0.72870995840994,0.2005882839459069,0.2891774697619392,0.002702430867422966,0.0002976904811151595,0.09688090157430468,0.03031394067634845,0.5538172808636281,0.6660953032600117,0.6497213725458773,0.6337929721115813,0.007787483476602129,0.05217052783801385,0.2438366008161,0.8250184945372299,0.003804035199473267,0.2421178908858981,0.003112693118010731,0.008790906517980561,0.07381227088837002,0.1029356334703967,0.2857221986137361,0.009244530256273968,0.007925228718774428,3.662883101351815e-05,0.2031043756051043,0.4204109881694076,0.2143591003854007,0.001277666251552855,0.7084333660691627,0.4315027821611556,0.06905144787489829,0.04035158618083675,0.07912717667097621,0.002685982290580841,0.6692234926441347,0.06102931340428516,0.387277024760424,0.1007053084644624,0.2439538687107402,0.6719633707733381,0.8260024114821645,0.0858970647868391,0.0005319661890763389,0.9790417841731053,0.01704106140466032,0.797043184476351,0.0708191289532635,0.8258060610049092,0.08452532534065429,9.5353638731513e-06,0.3728731988466766,0.006976122506429509,0.7716378402425941,0.001415674892425212,0.3674942657379322,0.978268423227002,0.9998867354594708,0.3347908322928776,0.2076003515874083,0.2323323792656682,7.988338371945898e-05,0.974586141396193,5.934392041554965e-06,0.0022066478639523,0.0053483021744998,0.001842632111503893,1.523927897430513e-08,0.8336221621700938,0.0164839031723488,0.7571995041524823,0.980730398142588,0.04045773600926431,0.6139746475177387,0.508071679945365,0.768085419553474,0.07362256013930718,0.5951546361533844,0.2107630520216896,7.966507547896278e-07,0.01372070163960024,0.03929298850169751,0.01617181715071887,0.06071976974668121,0.03484820522092563,0.3438884292321978,0.3122064376640272,0.04191427028279173,0.02561430083783827,0.003100212402498176,0.8448825728047383,0.5123694131439812,0.3277900852525183,0.103237813338271,6.422290292951179e-14,0.001107625408620942,0.3534599725325446,0.06080052485951618,0.9323579924296056,0.04755154220024749,0.09211454542895951,0.7855133917735313,0.1943925499150669,3.336818944800687e-07,0.7065790436814694,8.264034710766494e-05,0.967160322054779,0.591092588756965,0.01831562181638105,0.001846806614298348,0.08677779250340041,0.3688238974243059,0.8696042275903381,0.04493488614563337,0.8968125584915178,0.07709938492717801,0.1454529863829071,0.2841651980473264,0.1289986272787107,0.9444635942052373,0.3100805541592244,0.845129905717429,0.09824180898375279,0.3242199444506798,0.005382072185483133,0.004041349960744709,0.003667493289955393,8.117930473166135e-18,0.8197058301033154,0.5018730874998326,0.06519223199865599,0.0370186017763411,0.04203042804489142,0.03385576117957812,0.7923558570662266,0.00534956906432604,0.2595164919154243,0.8188129905540558,0.0177782242409665,0.5393439046301136,0.6892961234689523,0.01563278292851628,0.6206718567078529,0.6694548848197942,0.0215221184970875,0.4909227271028162,0.4540878693830382,0.001386558675451472,1.733407080802705e-05,0.1796664403982783,0.001983226733929673,0.01343488534289271,0.7319891408798221,0.058581422466433,0.9261881631239571,0.2474932975299636,0.8333668009331846,0.01873674459052944,0.7194545149350857,0.2274576584369215,0.2931820186135995,0.001312104206510683,0.01394282789291685,0.2834266513831705,0.0655749433822459,0.0001835014729351654,0.8393561668968763,0.08533575891055954,0.006973511712936865,2.019623153701495e-08,0.2872175259051807,0.2613689302714907,0.01135676895172335,0.0003125772012512559,9.549116741231101e-05,0.1342646405830685,0.7779375563785205,0.1435012008069153,0.1011277083950557,0.3581969906366797,0.004600343722380679,0.02964256267061269,0.08170665768584828,0.001100708550274604,0.0002219617037765126,0.0002578744148127022,0.0006916345256079956,1.539550136570256e-06,6.537436783314047e-10,0.09470816193206429,0.3377619622538526,4.878210350857657e-12,0.4057265749906372,0.8156839670805907,0.6284622700524307,0.3879196689937914,0.5814376389141395,0.001381071646704945,0.1065451521163443,0.701051076468318,0.8724625576665805,0.5569780421042256,0.1321654409835877,0.2804337502015505,0.1018542374056625,0.003353029916231989,0.08237825991811989,0.9504402949656738,0.1088519623931672,0.6906568482841626,0.5433786373534031,0.00295983607715052,0.1347052896028259,5.943782880946833e-06,0.2678386928642256,0.6279536236026422,0.4281404966974446,0.01680737322531172,0.168479034571986,0.008737653602988902,0.8329445902666059,0.08465964013641333,0.02416120075444589,0.9815400467257165,0.001293002520165975,0.1223373701381904,0.3061764770169119,0.979109083510965,0.7549197904039927,0.3068355314461953,0.1141243893133547,0.0002718085204641408,0.02083356474132938,0.2902602560198773,0.05456505265819649,0.3383769673888408,0.303674269675426,0.5578531456439428,0.001215586737728081,3.471848774718343e-08,0.001240799391364046,0.2503328884234515,4.406198082033109e-07,0.5792998088567491,0.5378397809596233,0.004461706837559787,0.9577639122661701,0.5887085069618114,0.04045579577302108,1.475914742281882e-05,0.4947075002767427,0.5952940931125715,0.5275536209935892,0.5155581262925216,0.3581939231518702,0.02073333283870197,0.0002742370530795368,0.5510495969831998,0.4797213122813993,0.4393816561878326,0.9504756076507352,0.7524196837813943,2.700915790930339e-09,0.03176419123158525,0.004186692441732214,0.4629780230292859,0.9097464855106044,0.3132671256265559,0.003050624855983422,0.6461473703991205,0.0003458037674307114,0.2397874947938625,0.145464139751515,0.1768300615298567,0.0944796903879238,0.003466099144690122,0.8553262667766353,0.0001707433151811537,0.6169686680500346,0.04117489904198744,0.09605631294586436,0.6593424851398726,0.4955386718590295,0.005673613217006131,0.1330218357777166,0.0009277469434376507,0.7084353397991796,0.2222444732252796,0.200183075139364,0.08503344950221095,0.6539211521940086,0.151364556445672,0.1526170321755389,0.570956496374601,0.4269626828752488,0.5036974714343472,0.8865419903642366,0.7031519967007904,0.08243244302166568,0.2841651980473264,0.3447303362058585,0.05491622472784295,0.1952564217605233,0.2442240629822304,0.04954079391822813,0.03460009791527973,0.6154086925051814,0.001654183763468582,0.01412607982746699,0.03195704109872479,0.146601647236161,0.02054390777180689,0.175084877969958,0.01447435134712041,0.02552113037747058,1.198608991256694e-05,0.3964751095272367,0.1208498294284445,0.421325656197256,0.09647462314839279,0.01918399058987903,0.8469562149725246,0.006919429236212097,0.385914465833566,0.489177313786564,2.745329548348951e-07,0.904232470463803,0.312717705494055,0.8706428981227584,0.9028804001080546,0.7002539600579584,0.252101242460937,0.0858970647868391,0.001550397600477776,0.01209237074023552,0.4772297134228393,0.6727045222851504,0.09337468790871653,0.05385942516775358,0.2371338828932861,0.980324187467954,0.8004536492826924,0.1029356334703967,0.005356129686997031,0.001278809169635206,0.255777146398861,0.3193254351370453,0.3589672050098316,0.006560028943563853,0.01476003380914483,6.80042954409427e-05,0.03335139335948647,0.7279406453547894,0.0001024583650935993,0.000296102986314455,0.6649793049293362,0.2518876483469366,0.5335248918443544,0.007002424831290665,4.395736144574197e-05,0.6802479151027938,0.03087875983369902,0.8121239635594408,0.004512695288665091,0.002125836884956651,0.924812934586142,0.03693887799562567,0.5282958689185843,0.001647790285004701,0.07489477915565929,0.726829551498742,0.3278922696108442,0.0007518440772880929,0.6117801829462806,0.002604408655253257,0.02403331481153059,0.5938465275893636,0.1649186693431066,0.6311911322870776,0.0188215490867183,0.886485069734975,0.1567864638683652,0.005583023073537738,0.08214945422429899,0.2281696009769879,0.1380590874748095,0.997885654716787,0.4013998043848392,0.01362610867003603,4.907896212855529e-05,0.9167416871882991,0.2093852344085025,0.617863207084248,0.5355757898521635,0.0187804542525606,0.01166405098916082,0.8816284484778953,0.6697996345221027,0.5441267241231195,0.3008546480017569,0.02261087516019509,0.5123694131439812,0.00482684643304961,0.02299493849493858,0.008726083042326915,0.0008864319292588097,0.003539989000976861,0.07775280659276744,0.007103695021252581,0.3675152285253229,0.8097098542241209,0.4275096955612907,3.14080230967855e-05,0.3687561694093667,0.9078770692133724,0.4074397087949591,8.217486727106905e-05,0.7901777693374957,0.003305926698942254,0.01850197907177924,0.1677785571605964,0.0247020157496858,0.4377222573998539,0.4468060751460923,0.6009182767180771,0.01033876410734699,0.8986391568818396,0.006986740287986577,0.07235590769082648,0.0004215540305256887,0.3530065253855647,0.9244551902744573,0.03069926676250912,0.009179029801177043,0.935058092661582,0.8904781998823377,0.1099142969885981,0.2163371822853391,0.2518815948198604,0.5853957555616968,0.02069119632447858,0.9603014331411085,0.001191836008829248,3.844659818672642e-05,8.268766317974051e-08,0.1846745478457876,0.1243978454290562,0.000140349549139629,0.7159651459700701,0.4840389970081971,2.081414442632671e-06,0.3609574111234633,0.009569206768556928,0.02878404116469898,0.6919678220004876,0.2784831394386866,0.9524919743482566,0.2838113983706829,0.006328718023225642,0.01065264841665101,0.008609736693208929,0.8575228383741343,0.6681761326770439,0.0003993493183087035,0.6929504880479856,0.4454536081701161,0.02445382970670921,0.3745763768844507,0.1459866360384289,0.6114383886835902,0.01251948093400304,0.03505317060828989,7.844641585592257e-05,0.697038916662684,0.06670935121604381,0.004473418332131941,4.326030631654101e-17,5.547296342584586e-08,1.645685955412658e-05,0.0802906586598483,0.6734434660698077,0.7109721649904116,0.09780975692064302,0.178374036330271,0.0009740897816998184,0.2418929417485931,0.08357870025715192,0.1280389394054104,0.1651590083184669,0.002555505488364565,0.01858304876860371,0.1745273561214586,0.861860273558655,0.009633788831384353,0.2835582803953587,0.5700197427365824,0.007242174989327292,0.0001806780307939371,0.6820693415144117,0.2198249428530617,0.3608126149729783,0.9427498489586895,0.7383365371553285,0.4744955800713105,0.4843694387382838,0.5478076390052616,0.0007521768122751422,0.05871522205877133,0.1734834882980639,0.2688071119130441,0.004161714510503125,0.8900179546033162,0.912164889569276,0.6571992999212716,0.001345313521438893,2.043861241582043e-05,0.3979622728088988,0.01839006815114813,1.197486954082552e-06,0.2666386300672767,0.1374434800832936,0.2066837379520569,0.2091397866523438,7.520544926659862e-08,0.538193880039897,1.074942055659154e-07,0.2739270921305027,0.8900179546033162,0.1171991489866005,0.003467493532813639,0.5185617890942578,0.009890489819050838,0.9577315007099367,0.5417844250867309,0.003224386178406124,0.1567346802910551,0.237016902345304,0.5279903579026678,1.695109282226911e-06,0.6319745610229232,9.849854588271646e-07,0.6253921959554808,0.008065745236318365,0.4851666739502515,0.8135232163197789,0.9721707645220129,0.2922916032664845,9.094482569834947e-05,0.6459307649583894,0.0001009503531603262,0.6577189267147815,0.3244802067091578,1.85222815814763e-05,0.01147308193765704,3.748670377778753e-06,0.9518323344757121,0.8902321277651541,0.5510495969831998,0.008858966177839904,0.7253641196711846,0.03416778308697151,0.4772064183405466,0.001505187146714925,0.0005406958221652452,0.7441391648846232,0.3661288415036142,0.7712848002975813,0.2638074379501316,0.7413732707489581,0.08958465477745048,0.01574655220248182,0.006720628512003459,0.02661674840552994,0.06209398080941929,0.823325793006907,0.05156206041248487,0.1169742941402751,0.08698056494841924,0.0422986862647885,0.02096923337817039,0.000623562426360559,0.6932449873831832,0.08908631777468663,0.009797683354513378,0.0007446012379844788,0.02658603148688265,3.262753844361592e-09,0.03124647042345548,0.3084614770411561,0.04123006730127633,0.03484156320253827,0.0005521413032445327,0.1445155970984781,0.01770684889770349,5.183938947491446e-06,0.06039925031148648,1.589648368088906e-06,0.3686301706091279,0.8518302758896724,0.006307928277899667,0.8415730103256502,0.09418518868805047,0.003448168705459314,0.6421562566562343,0.5614482377319951,0.4182465821064267,0.7306168824037711,0.0002506873633525182,0.01737217317993377,0.8561612690601699,0.8217503979348855,0.7685965262254677,6.043409265307203e-05,0.00223941188553341,8.743873428956436e-05,0.07025620239207593,1.180857831861327e-05,0.1385875001550627,0.710949334101374,0.113656684686888,0.007037781183847268,0.2026574263912757,0.02865572857850524,0.004950908504215084,0.0530357948542042,0.2479724464726876,0.04070620617501393,0.2787548853807957,0.1785711633599749,0.5892357138780376,6.270399032496997e-06,0.01803401426875245,0.6705655673559546,0.2149689761070692,0.003944477817369534,8.731900663001084e-06,0.7398698243078285,0.5605434903725856,4.447096427073036e-05,0.002222552904867521,0.1135273734411675,0.0004458143614909128,0.002128110496308547,0.2910720222068587,0.1755989717884361,0.195438006040356,0.006249111330587902,0.004643141072744701,0.09605631294586436,0.002579281061954985,0.6476482190244769,0.4546819755429568,0.9774412545632509,0.005265217655995512,0.0001295006722720277,0.002528141593467673,0.7745327958319056,0.70558544842823,0.1602918555308364,0.03825796676602171,0.3116321976085482,0.03486405397246686,0.04436506747933455,0.1909545179726584,0.003489098765096567,0.6111272779236127,0.09576318023199985,1.658482634585585e-06,0.06365572823436599,0.1949805582258884,0.1009469824535854,0.1630859100223507,0.04383801548226376,0.09456025904973937,0.1569797400861699,0.1337965730736302,0.4558354515034323,5.495476490248281e-06,0.1108257812864162,0.04308997346163529,0.05643265580275394,0.9844723428175095,0.5482236195762433,5.104375276392988e-06,0.01255306797702727,0.0001469078604997747,0.07017482526987234,0.9790417841731053,0.8580608703686767,0.8309675909325682,0.01199930983576249,0.9755630258724766,0.00659606120617138,0.005279974753901392,0.3193254351370453,0.5911709982901304,0.1518716503080129,0.001639300883502029,0.001074707608707857,1.706912292142524e-05,0.2188695135251888,0.0006620944593988441,0.04895268987820338,0.5382089184983057,0.1573607362667652,0.8692619594240978,0.7715267148079097,9.513474443843383e-05,0.5443021759638934,0.1207370828100211,0.002582665830559923,0.008043965650617123,0.0002697475828319297,0.2918371260882386,0.2172135818056451,0.4191267280504116,0.2390692537968785,0.4727328083309673,0.4872804810065125,0.01721355606497938,0.6997554511405589,0.007426164180613431,7.610626806818451e-08,0.01397999942204175,0.01890372343458801,0.7689714695996296,0.4738617222534322,3.754021382698597e-06,0.1254659122994257,0.04235830823967331,0.0002867243214627624,0.001523669641865164,3.894849472788616e-06,0.2296404012781049,0.6840426059458385,0.124433215558786,0.2877999980209022,9.024639565420238e-05,0.1822036597989343,4.267855232277573e-05,0.2500421443148282,0.07605346796800846,0.4471802680671846,0.00390592178469176,0.05385687490705184,0.6116990012562902,0.662778416494866,0.1243469750129727,0.313766656847133,0.1994664727128714,0.0004653963190845811,2.425852161271012e-05,0.6773855046530396,0.01856576469564138,0.0005475614526568329,0.8957634604792314,0.3206070390201082,0.9731099902041823,0.980564956918433,2.630791896325267e-05,0.5129816046192556,0.007707073226050807,0.2629155135470747,0.8116145193105934,0.4123276391321968,5.897698266410722e-06,6.034832613667231e-08,1.4086484158161e-05,0.3609709851500767,0.07678808489591812,0.0002533380621561138,0.4046234114889801,0.9060020163937786,0.02895140473924024,0.06478728335520029,0.8467230836712114,0.7058804036212849,0.3788625730273884,0.1190556023206195,0.0003392393140707998,0.05151029071132131,0.4245644849051275,0.1722217424064633,0.429309743448018,0.8689828680530113,0.4932021797326748,0.70558544842823,0.0005029308032898218,0.3610737525174104,0.002345061583879018,0.5683046345843966,0.1340813508218436,0.9069463543390744,0.7262184188444725,0.38644517542041,0.06255954945607742,0.6906543089424633,0.1444299157183492,0.05335903543215022,0.2058654653714495,0.001934619323212639,0.002471488358794815,0.02316965494443643,4.581448146265518e-05,0.241203257129888,7.045864869775181e-06,0.9869364783357176,0.06747783581974048,0.8208417973651765,0.04735042829536854,0.3107547020609976,0.528460762535179,0.1408009728387026,0.1840592460378929,6.023580455518079e-06,0.972020949177748,6.026435856414606e-06,0.001800568165888119,2.293983708681851e-05,0.8080565694956535,3.344857482458638e-12,0.6405766724402243,0.4262098099081066,0.837678263996971,0.0002105764455261702,0.3812677956344574,0.7178735349320836,0.2584202985767459,0.0001525174285639355,0.001171444717267949,0.2782839206654715,0.0007067824959959546,5.17089448004061e-06,0.4765173639594069,0.1934736865011907,0.007925228718774428,2.212203283047481e-07,0.406951017706053,2.831043516929595e-14,0.09027268516679969,0.01886084339992112,1.232228607403705e-06,0.01362465055561483,0.2127851552877829,0.0331772756310862,0.002439409285323033,0.2213073289748718,0.000897876283347662,0.02544153404684202,5.639919394563182e-19,0.2769158355252839,0.001255446271814683,0.8845042095422153,0.891106181570491,0.1284650259501947,0.2949078137519708,0.894619521416063,4.176929134171678e-06,0.1878316402522684,0.792735655547172,0.3718745186090832,0.01667718548920978,0.09651232099499769,0.02809515645901046,0.09672125096671452,0.001950859212750752,0.003750700770603787,0.6037081713483419,0.1165170090793727,0.2395648003504091,0.02219615899481103,0.08477213399202969,0.008264844389604602,0.01297855415269972,7.679489025753749e-20,0.2854434214358612,0.9996962035915474,0.2005882839459069,0.3824945509154183,0.001106985698197307,0.08119577358993281,0.1624754047880157,0.003828318132032211,0.1218525610791666,0.0254498088844656,0.1000236070832623,0.7122863142839156,0.02651241338889049,0.5184662018336651,0.01647415757027887,0.1317248269473891,0.7565062201517988,3.637454225763887e-07,0.5847007783291761,0.03411276925477202,0.1695494732021268,0.001274126366580263,0.322757051963168,1.438406730146919e-05,0.4531786482051682,0.005964401317693692,0.0002356820847316189,0.2591055893144781,0.2536108466898382,0.09230973168228518,0.958434954909163,0.2632344958028394,0.3001560385554676,0.7582723710703365,0.4688758684401629,0.0001578390836450777,0.9793259069237565,0.8930210967687493,0.8200478747596079,0.01537389107986435,0.4595784399278397,0.006900242125443201,0.1680734831816569,0.2511047891679544,0.2438618255809196,0.5122364459920586,0.3955405371362141,0.4123379756873115,0.9903576758265026,0.03150723823293047,0.0001914046835427498,0.001517624664768978,0.0236574237964063,0.3795311151071906,0.2868482104298517,0.5810604801569503,0.4032751063593272,0.1846291844397388,0.2582575719075826,0.006220895673199392,0.005392611162973988,0.5131757373211651,0.009637790144421969,0.8304129322816477,0.2106856050076767,8.067158630125555e-06,7.240427164661696e-07,0.6900355111383188,0.2865549831025819,0.6350365973353086,0.2887077607918507,0.5941367288202518,0.9340646862996124,0.5949380853459836,0.00592133178030131,0.4060194083390927,1.448787257941782e-05,0.3641145303917463,0.003566791741208044,0.9279533416194785,0.04449915682050216,0.04763577770787303,0.7276449219796548,0.2702269924993373,0.01989548739779752,0.7972083066375606,0.92273907670411,0.4072263055320604,0.8842088970804118,0.03086488182241179,0.0001548007419192402,0.6356634489808461,0.02686381057576108,0.7183176905062991,0.8035012017909385,0.01303948791456496,0.0002258294279759841,0.001280253868007209,0.4651688205265315,0.001279016266523628,2.940440693884134e-05,0.003796918568286903,0.1616409818076852,0.01380789961268168,0.5087640325179282,0.005339690371195848,0.08732341391952941,0.7549156811384639,0.6377276338816739,0.4946625691118439,0.1031249298997788,0.9052182111585637,0.2544045396343461,5.039420886501699e-06,0.2364323863185821,6.166428650902677e-07,0.9133013674161937,0.01751760182320606,0.002458616875840267,0.0009558526573433395,0.08303267364981973,0.1079258649907685,0.4643969239524532,0.7310216420855912,0.07187848936845467,0.003481732519668392,0.000253319707713084,0.002121975729709717,0.0004508081674817186,0.1440632688210531,0.768085419553474,0.9641042548663927,0.7813552650888332,0.5967277656411298,0.3288948737724424,0.0002310952144165096,0.8218276621854582,0.1718475385428015,0.006720628512003459,0.0007401170525645239,0.4459447529764113,3.222132761592621e-05,0.07464331880562607,0.9995587242417702,0.003796918568286903,0.004265287369211639,0.928353225344158,0.2367518621420053,0.1952531405832232,3.819773791815423e-05,0.891131492131317,0.3211924639291927,0.9538898226702879,0.8046152331094575,0.4693506968568273,0.00494540435240398,0.5730448390910953,2.534839707901582e-06,0.9342125877308195,0.6552497994535325,0.2746062141368557,0.054580198455988,0.09557530006333745,0.3872696441480604,0.5609205840332261,0.2173324761206944,0.4604065627750782,1.758739286009983e-05,0.9939109395728927,0.0632313464865127,0.001653082703141432,0.03741159315136845,0.8940376292345515,0.01982512507101882,0.5217012593723287,0.5544718075265195,0.0064577405404811,0.4231890800811932,0.2728619597348758,0.8811348616346973,7.067325869030802e-06,0.007900884442279234,0.3571505912406167,0.44926510136291,0.6429379618829858,0.1787093184124466,0.002582665830559923,0.2706511068638917,0.9581384024681207,5.054237466274071e-05,4.767743596158191e-07,0.002528141593467673,0.7087266231513016,0.04130910065988014,0.008737847684229222,0.01029199662270448,0.0002869399045044566,0.5091199361905344,0.467744220665987,0.5386945957132041,0.02273117285354411,0.1302923224717469,0.227892846607932,0.1589775907517313,4.14950087481101e-06,0.3795584095594416,0.004115823916328719,0.0007043775730569495,0.6351943460337925,0.06428211804322413,0.1161633205939835,0.07773376132960608,0.5414620756962666,0.6246357500878706,0.9907340021215861,0.8900179546033162,0.003870828089844828,0.0001044916203667703,0.6104592712407766,3.113614043721823e-06,0.02170771150948655,0.0003418636254690881,0.5539565520680549,0.3350765525584663,0.2538751382975648,0.3796695180332181,0.4690411666352498,0.1025132654018018,0.6738341946419159,0.0084145921977626,0.04776408835743593,0.1713601774049633,0.4873049990213263,0.192045271957477,0.5543363277401123,0.01146567537428433,0.9177911940891246,0.4098598342599275,0.4772950107946017,0.6813679277809334,0.2314038288895108,0.0004776385865821503,0.03649930875822276,0.0001098116834786642,0.5614482377319951,0.01852238784039248,0.904232470463803,0.7815576867706236,0.6283996956987788,0.6769394981142588,0.3934883122035482,0.5369718588302902,0.8165059634500709,0.04188197891775073,0.9107085811692675,0.7769166684898277,0.7767080546682494,0.1269507423070103,0.01968744402259432,0.01171748867167927,0.4230620196499208,0.6921877607975235,0.3173998670202456,0.2691671675849953,0.1743876155692449,0.5370899381188965,0.8900179546033162,0.9091402574384316,0.1838805410038354,0.2885688618990723,0.002269770729408341,1.58603464522744e-10,0.3672475262888054,0.3953945210014464,0.3145634916542609,0.2249335704860138,0.1722071056130867,0.01608169434084378,0.01897617471746292,0.516593740540374,0.01372316090322501,0.6681761326770439,0.4268392435960078,0.6169686680500346,0.5965457162195456,0.5052662152273911,0.9325790090528814,0.03090109957942759,0.006076662613560848,0.9673480883029281,0.002760694122013438,0.7816851519491024,0.07053627736497489,0.8475762831286417,0.6012187622103539,0.0009906806031977364,4.249379768908312e-05,0.4757006466850444,0.01987413754880617,0.5775936182589401,0.7736578529229305,0.1652618036643446,0.9846843401488846,0.2595152319885118,0.02840830424403104,2.564582768393014e-05,0.004017583986093295,0.1127993027758567,0.1317194767145437,0.9154511691709356,0.958434954909163,0.3135879373685328,0.5790302206871186,0.06190347787174207,0.3848333277545393,0.563654875639284,0.2064414732378868,0.05433374650102887,0.08418536845222724,0.4097753319037919,0.2087911992746692,0.1484878687455582,0.6159732576643792,0.1041616005518773,0.7116163875941875,3.333342706283794e-06,0.1707957353705236,4.087681268975317e-05,0.0687083066719453,0.8102923062457426,0.6590098999742752,0.8332937900089283,0.2462478174259664,0.50346629193641,0.008208120546505463,0.3044039131111565,0.3773395480590458,0.9718452190424115,0.2105834292216518,0.07955890371153386,0.002947281103538909,0.3188903113214494,0.6914845800927206,0.1938208348073516,0.8067752574346339,0.0001048716419390473,0.9943072638563226,0.6524212104156987,0.2007007748565381,0.008858966177839904,0.1067176877018728,0.5027618446430968,0.4836684503654015,9.819543027745383e-10,0.0002433804677414921,0.4562735102239706,0.02141740530847507,0.001480147677027225,0.6122515804537189,0.2769855543816727,0.0744227538527307,0.03335139335948647,0.0001344914444730803,0.3656812432662664,0.05933608745697724,0.1239977259109246,0.1517904201121119,0.5903216018403359,0.5999895952100606,0.6174720198677234,3.138200152690684e-09,0.02363023418727694,5.320024800650212e-14,0.0444697275348151,0.3432245332312852,0.003077901284368456,0.4667708626376122,0.2996047872231209,1.107559731900218e-05,0.8099388082353778,0.8368426856616589,0.3145740675086033,1.153695108764015e-06,0.6865303866721716,0.2285416371260276,0.003175300097766335,0.1649244489379334,0.7913946718773649,0.1069795855144226,0.000435242829678984,4.679116740372613e-07,0.193176053410852,0.8564610357247064,0.7944818460387132,0.2786247365147359,2.276020283583355e-06,0.4678378048116661,0.1350412300336289,0.006545905626416625,0.03201890900449889,0.02147528067858338,0.4230620196499208,0.07683840332493329,0.5248999841230851,0.6478448062432947,0.09276644018847098,0.3046655010094583,0.9443736495084268,0.3495101552384121,0.2099969565721888,0.21101262567968,0.5784747550347176,0.02651053077340777,7.484730499756385e-10,1.124100047679905e-20,0.421651573819744,0.1342349479596148,0.9844489058736869,0.8957685178971093,0.7704780230516841,0.9888089540461193,0.9029994061935146,0.08549071049898652,0.6455913491277118,0.897291749804881,0.01528659799423745,0.6523300869301871,0.184704262530644,0.7453958782589798,0.01959805373153822,0.05240150738123754,0.3028884741991681,0.8900608928127817,0.358670275879795,0.232325613835414,0.8174652706922432,0.04372697045383021,0.7492320833967557,0.01029465662864775,0.04315099550487222,0.801707004782642,0.000711510921841876,0.08568442966383667,0.05030563031791797,0.6078504176607773,0.01845500976306139,6.928097972374971e-11,0.8981768256616506,0.6001828628146643,0.8522124992781847,0.7449261622648096,0.7641975331731772,0.976566286178018,0.0002757343294648798,0.6018016149436242,0.6167427945919683,0.002696624290387607,0.02673544491260007,0.0006644647085048089,0.06344636711943984,0.6612940341656897,0.2474932975299636,0.3340308294173013,0.1518046159881064,0.001257594571533946,0.1245206890254876,0.9300516736671156,0.000676397364724293,0.4357964659439116,0.1032084118166321,0.9423189960178995,0.1044014605298139,0.02750366510718041,0.5656483984185401,0.4955901636638003,0.0006748507876928551,0.844400565870377,0.8879456516894991,0.0006237246967582409,0.07182236180818292,0.8031677058165605,0.03482348991320424,0.05978089563452722,0.6419755740197174,0.1282617389817704,0.08584988175635677,0.002184589498650554,0.9387497473397257,0.3381192411374873,0.0007677264384318437,0.902315967789127,0.05356737028089055,0.4649436058541061,0.01764285668423373,0.0003142932956242839,2.738342618682541e-05,0.1124988692157867,3.706610738579053e-10,0.003669605894442058,0.001331308086964639,0.8898554685903555,0.5614393799185037,0.3653281496956803,0.9441642991323108,0.5522741854482243,0.7100369393381861,0.3837466323899458,0.8995150201323119,0.934389916848643,0.9091402574384316,0.05795724036803942,0.1567346802910551,0.02345576020824755,0.8002558093062375,0.9060020163937786,0.0009112455380779819,0.01681203668378827,0.7408635786055862,0.110422123293955,0.005638814247062336,0.2131192080559015,9.587097967118237e-05,0.6048375847457848,0.001617149153313026,8.526081801991859e-09,2.538112117482542e-05,0.004813236379092207,0.9995587242417702,0.2313973315475988,0.8231680191060285,0.0853826199485252,0.6389731408085969,0.00763739916897103,0.001607380931172892,0.1464909361707261,0.04970198452346459,0.1600474329783209,0.5542814505267466,3.709854012881483e-06,0.2009130111024074,0.958434954909163,0.2214537179994338,0.01648780940343677,0.3990166100405467,0.2087911992746692,0.226710797947595,9.729474656317405e-06,0.0003669306775564062,0.0337046565141634,0.8794010271605475,0.2245269075382025,0.1424420325323898,0.551157586676237,0.8594777787443703,0.2577145436742577,0.4039566158383088,5.304425033633682e-07,3.042527583539181e-06,0.01393410375762854,0.7167699788084732,0.07160813434389091,3.357097965406192e-05,0.2499812322679177,0.820098560440222,0.3543314645885247,0.6980063172564887,6.459641956561512e-05,4.247934934904894e-05,0.1363190328000018,0.2717207962351317,0.4862829221924109,0.002085252038876735,0.0001616928844152857,0.7624007791915542,0.03374774468121499,0.02262553508704787,3.671658410101672e-08,0.2152211184149358,0.006632632573841018,4.000115735611104e-07,1.663943657234395e-07,0.03343711727728113,0.3761267866278031,4.521715442886486e-05,0.02124890489607559,0.08257787728839401,0.07633894735983386,0.6549701309346277,0.06076556515519627,0.6914717116674132,0.001095496403373738,0.01578856228781265,4.06504550965072e-05,0.9963563960805795,0.5774732878916822,0.4757521436334332,0.08431756347103801,0.2104085973654205,0.2527619069945157,0.2182891438810337,0.8623279674327174,5.675230070475227e-09,0.1729520800573019,0.9524919743482566,0.3556870504712094,0.0062993233863459,0.02565651016651843,0.4419521434325333,0.001074417412260975,0.3643187224903851,0.0002432230460388119,0.003363631078001442,0.7396168731050784,9.46306664253986e-05,0.04525058664819044,0.03141145714332307,0.01357135607628672,0.38644517542041,0.001417708759507125,0.05645629408325904,0.0003020733355586088,0.2107630520216896,0.874664028679611,0.0568965244907855,0.2447286692703358,0.8659486770098307,0.7384660243779744,0.3017219230132846,0.1002473659116109,0.0002116295770344186,0.02771061045657783,0.9648598480122718,0.8890104304153927,0.2121897873629459,0.7746162828844936,0.008071885522646953,0.004835597102945544,0.00139604457182405,0.6977103769976428,0.3876929978116246,2.10524594325566e-09,0.252101242460937,0.0008169222643398387,0.5058171370863256,0.836518614933368,0.2066458756472906,0.8730132228327471,0.001512364303488909,0.30379692084183,0.01305435780171323,0.03967693399462542,0.3163501129762324,0.005539763784481781,0.9581384024681207,0.001540704360016849,0.2646573902338544,0.5226267549415042,3.423079753692918e-08,0.2221359950797269,0.1044014605298139,0.3232873696240325,0.6955968312277975,0.8679466454155162,0.4230620196499208,0.00536531512555695,0.0002686011778850215,9.677251328444869e-07,0.0009699966765157326,0.5878666360280891,0.1986421645848077,0.9862044037773837,0.06284329944248425,0.2014424711501141,0.001458935335479238,0.001797618648985034,0.000219830078570694,0.7061023607995531,0.202943940432713,0.2577145436742577,0.003767582784075373,0.8714900036831484,0.8205516754491651,0.0743804941334518,0.1200978288166288,0.004066477950110879,0.001499781526833245,0.822710270261539,0.3610039133868509,0.02555033442327474,0.02584423025339925,0.3278161663940747,0.9485196981102475,0.01565779199420693,1.469580236028094e-08,0.140161118198498,0.3517035730821587,0.02327535516789108,4.134869569379778e-05,0.8516649070122478,0.05546948697602725,0.2740112349503517,0.0309339966755749,0.6571992999212716,0.380680083786468,9.464795359290904e-05,0.05867457752917067,0.02565930448235309,0.6734434660698077,0.4300552965528582,0.3363744120636083,0.6929504880479856,0.5107584232883569,0.05171168726172844,0.02116954317054326,0.2864068204501525,0.3188903113214494,0.2940895576003459,2.339351773419847e-06,0.005771089579970288,9.32466034584964e-05,6.342221446163807e-05,0.1598239608140606,0.4865837926593759,0.0085855713718239,0.9100610035416831,0.01468196220173346,0.3931949214665691,0.01505441814666458,0.3948784538424147,0.08471070692120948,0.0001688041653541776,0.2834266513831705,1.067599123276048e-05,0.005011668826860729,4.553426969442089e-05,0.1017628605776005,0.04321410718363337,0.05850455089637858,0.1989583797441934,0.3095632823974481,0.07023203033423701,6.130395936680965e-16,0.916178359361523,0.00278538944793735,0.03967323370265666,0.738527963265101,0.3616420770176487,0.0009878755582696504,0.6498399416086346,0.02273723237062232,0.01769623331405449,0.7160063945852628,0.306088692303122,0.3335209434087815,0.1845798444462507,0.0003535717313345773,0.01801726356118933,0.01223920658110021,0.6001045980366944,0.001384804873150879,0.1795482113561157,0.07518301976933968,0.0002369097512529797,0.03096206472572652,0.6275119726763164,0.2231134982955826,0.1589648866484757,0.0002365043607279373,0.8832375185852381,0.2091892334766048,2.43309440017806e-05,0.6577189267147815,0.09581708483279557,0.9098278533952394,0.04375157597661909,0.8624803431830187,5.137098271324727e-06,0.194750817341081,0.04516494404574694,0.5905007907756435,0.04189074407139834,0.5872461103482735,0.8338873806120253,0.2198212566313772,0.7698198411563923,0.1367900657069512,0.2175922529985906,0.5292202583335528,0.2073025501865403,0.1583946757729478,0.6931523408889417,0.009655795566902162,0.2107630520216896,0.1010609827382184,0.7819864358074724,0.2512155714566888,0.2419268691033135,0.4101924920971234,0.03333869959278044,0.1067176877018728,0.6399646661296903,0.6350365973353086,0.677273085188457,0.8523648033998623,0.7316124218597773,9.496963227340618e-10,0.02631264119761623,0.941755520328858,0.307984787178276,0.2660705610152779,0.01909676749638499,0.03859817615233522,0.7161057205547999,0.140765753760884,0.03044404534011368,0.07645339536837277,0.03345176212518308,3.190830572995348e-05,0.0199773020919944,0.03985730020981625,0.008400505729225972,0.0004788470035945707,0.5551414806356442,0.01136058664002918,0.334049029508252,0.8239079261731828,0.0236455766733563,0.1128974612775209,0.03507718541613113,0.009548742025700566,3.348431610740621e-05,0.007757721715324726,0.5296725361839201,0.1200978288166288,0.1795650402859425,0.0003853570137736705,0.9995587242417702,0.193192181653711,0.006964988900611057,0.2194330213589155,0.9762479590070344,0.007285799134634661,0.1031280737812602,0.03050219296591155,0.9274107704819969,0.1750918849361759,0.001687066362387199,0.9296666096680491,0.8320955062226633,0.2809292279087133,0.6198146201822144,0.06774191043777424,0.9121054364682473,0.2120009792148984,0.9145085055949036,0.9296162245967607,0.09557215583438576,0.1212300156735773,1.80597478245666e-05,0.000599909169382573,0.02875183788637119,0.004673300348646076,0.65805380259008,0.5732359037211383,0.5505346200146731,0.07508379750161707,0.9762479590070344,2.550164429783031e-05,3.845610894725182e-05,5.788389795973474e-06,1.062535946381425e-05,0.7867340190814948,0.3453844941170415,0.3093306380276906,0.1748150936409668,0.009791506000075509,0.09605631294586436,0.1447591865832703,0.02878404116469898,0.6590773652780622,0.6588857542065193,0.006220895673199392,0.05319139659643551,0.002833068046457967,0.00183596112569291,0.2605961960393389,0.0001270290914355896,0.3263420325899197,0.8616642987357883,0.005885320493867912,0.2613689302714907,0.04387982520804433,0.7005711754717471,0.0001718658851900953,0.6704628541423342,0.5521251202699506,0.1392608237036904,0.7025468519260948,0.6369097748465854,0.0001643687673026503,0.6447659665917317,0.02363023418727694,0.2996047872231209,0.3943409424481119,0.7432042668638392,0.9996962035915474,4.084115781821167e-05,0.1673003024724393,0.0302995812217828,0.296297077550379,2.774653888418531e-07,0.0006121561984619267,0.4154366831019414,0.000203471480882368,0.4370089936682969,1.42885104037535e-06,9.308429317669935e-05,8.310360371518173e-10,0.05960646555155347,0.6562002579987023,0.2194453904220377,0.8714900036831484,0.2205547117796461,0.3414203850830169,0.3188903113214494,0.04647580263190117,0.9736941436714664,0.008024456012243665,0.5823484499920405,0.1735725494317039,0.1216699414909406,0.04787407762782048,0.3416381642005724,0.05156940251772334,0.0006656531448745498,0.6427269979018091,0.0003441011092295611,0.1937954708161001,0.1522156829025113,1.699869846766803e-05,0.6279536236026422,0.3593630278710753,0.1177813380192164,0.01381143857842224,0.333356080509241,0.615726001751347,0.735312514734545,0.151364556445672,0.04733185724811995,0.4513446260658638,0.1303754076590176,0.8616642987357883,0.9918033650321997,1.410941026483532e-06,0.4851509326618199,0.0005674005354194731,0.826992147773028,0.4764026876734136,0.5728484855246055,0.6485379760619381,0.3876929978116246,7.557395836716659e-06,0.05578955197192532,0.1759787935607808,0.4910423515835984,0.01431742715339074,0.3752628094642079,6.991711466071168e-06,0.6095404457380045,0.4433267086368337,0.01401362515374122,0.002693580755493397,0.3228041971558721,0.3230995405534617,0.630901211093713,0.007731317260610836,0.09571353340786509,2.949408240893464e-09,0.0236574237964063,0.0367195988357507,0.07816963487953042,0.8655581733395452,0.01273501291019739,0.687975167766661,0.1207897126843202,0.06230455098335568,0.7154167343906529,0.2749873264692645,0.001788135672007327,0.1367900657069512,0.327099332061344,0.8981169435964241,0.1178023469734934,0.003044008526785121,0.2898648828720929,0.252101242460937,0.001277802697660845,0.04349757759481648,0.1664847119615278,0.5814468440877952,0.6348256990293504,0.4949271420670546,0.2212947605289362,0.007712561327073252,0.2521611258031755,0.1326989467635156,0.1697893764766983,0.3988390077615484,0.612078830631426,0.3061416174818337,0.4188713275447348,0.409142071484859,0.006357375151616038,2.975008234739505e-15,0.1318790906764331,0.01416770561599104,0.1060253362782586,0.8772164942573457,0.2992035617972915,1.092865337774608e-10,0.9767502654995477,0.002935112566391629,0.1029356334703967,0.008737653602988902,0.01760237641815095,0.01630942155955275,0.003424682895224636,0.0003213889032017145,0.07412343478887823,0.6025176778739303,0.04717438672591211,0.705283662089433,0.2748615168008361,0.2933884825668035,0.6562002579987023,2.963267628457047e-05,0.0106611259355938,0.3497752736009602,0.05658980509494402,0.002032477460844972,0.6906825142042534,0.8115939446538615,0.2397089099873483,0.87737990444522,0.5010913414891739,0.002041714627690516,0.6427269979018091,0.01046928365039541,0.2209115854883318,0.004387406337303348,0.5647243433143074,0.1559173261612475,0.9953662136556667,0.04592044264058315,0.1294891425251835,0.03897871243623781,0.3829251164880234,0.5853909277672918,0.0006555186539261628,0.2353418382068505,0.8714900036831484,0.1472447516725917,0.0001408939066058308,0.8073394219814963,0.007164699200807945,0.006884244710038513,1.075553339852142e-06,0.7129517601702192,0.4443903297451697,0.1306277585299884,0.01097361160245968,0.01628637343524256,0.3312537789434175,0.04485725212356819,2.424080803785106e-05,0.48974898852165,0.3188216805619071,0.1239471745250098,0.06272597719491771,0.0142994685494714,0.03122731081213246,0.368121221481871,0.8667170408040655,9.494532671907164e-05,0.9778334487304088,0.06382119500910058,4.856099123258546e-11,0.001061543291330408,0.03837932682729837,0.04813810628352352,0.8873238426733043,0.6277636481359601,0.9068061070785892,0.993038854172052,0.6327159938122524,0.09795840473150112,4.211260865529816e-09,0.08549071049898652,0.09913037331005423,0.0001661738776496067,0.1042379016071077,0.05282558902641996,0.5020616785353801,0.00566440651168283,0.2097053213369122,0.74176862680041,0.1027189467016376,0.0009221562563660406,0.0006337099881281482,0.3360269504515609,0.5057975062320931,0.9260135068371551,0.7841610591431083,0.1093879266312035,0.7637641369585393,0.4914105525305876,0.2671953346533345,0.0002155079478086372,2.553680870841814e-09,0.1874431558258613,0.5052662152273911,0.9429326251164324,0.02206376155658548,0.5132276904010293,0.03186348136458031,0.359158767394255,0.08016843880616709,0.5004490191695895,0.05201471084311787,0.02788535346422821,0.07298382897269347,0.04981229699465892,0.733454074067197,0.4698974611451645,0.2873144722258079,0.0009277469434376507,0.2656697162915225,0.5192235235918178,0.01142281017233802,0.2024650968800027,0.4948968101483704,0.4652991505994641,0.9844489058736869,0.5554209379991504,0.2745133054664109,0.5223873212775242,0.1201288989303228,0.07891140639423345,0.8822691682684414,0.3393204183836807,0.0227859415070519,4.970546567312518e-15,0.2792127076466141,0.9373755546466254,0.9078770692133724,0.9524919743482566,0.6170831324447729,0.04409545866541729,0.1769093949449015,0.02508076038187467,0.6612965781712661,0.1142561768309885,0.1958254036685794,0.005252137113310742,0.9610596824299972,0.06004494455168718,0.4186998974002583,0.3678772130299327,0.4278286323695806,0.7074084901978238,0.8965597983968053,0.9815400467257165,0.0002081887944940186,0.002690971259263763,0.9444635942052373,0.2038504739417579,2.240821638458156e-05,0.001134334684211873,0.7287285621260873,0.003484954384706369,0.1921920391176188,0.650999192346194,0.1934736865011907,0.4242863564631523,0.000517715062104542,0.08042515974539252,0.9772277390631341,0.0001653492696876185,0.9377296281998976,0.08249698025859215,0.008579488391880337,0.9156949331109341,0.3605397341517651,0.7355283495349483,0.682319966210984,0.03127570244539716,0.001665896883372897,4.682003416786582e-30,0.0006486780168207013,0.6771116148067291,0.1852528141762073,1.606817481873157e-11,0.001317902713393793,0.531240235136886,0.2999478703511758,0.1031280737812602,0.2331523311532871,0.4155445691769533,0.6914717116674132,0.443764676075021,0.0001112646238640057,0.03729276125522654,0.6399646661296903,0.002906585509922045,0.6253876628777095,3.751482112454917e-10,3.67356607162019e-05,0.7698198411563923,0.005415164556192533,0.3210589421711309,0.00183596112569291,0.000103510768485004,0.4243696619766324,0.1193911624832344,0.2658948872456393,0.002499319474210117,0.001207700120116115,0.6198146201822144,0.56028576112992,0.001656869995112922,0.6529374910576278,0.2076380332871269,0.6388474998087332,0.8763313836932529,0.3488614770677054,0.4411653577834814,0.3809250405798698,0.0007401170525645239,0.8346695501487332,0.1144397998200778,0.2245745171046289,4.130864440254806e-05,0.2279643201516018,0.199554498571902,0.5386056005593556,0.1869096362727519,0.3672192942066637,0.02124470712294015,0.2315879054109945,0.6734434660698077,0.2421178908858981,0.9109177252497807,0.8812803241715809,0.5396109859119631,0.04143815045163816,0.3178163510324212,0.004147101624124841,0.8144273385661703,0.2655714961148333,0.2320759592356203,0.003450370587126521,0.3812677956344574,0.4092150823699344,0.03665319619058243,0.1003660084869968,0.6353072062146693,0.2045200610054266,1.549839093825588e-09,0.03524304077205923,0.1680734831816569,0.7446221204799977,0.2525388579861339,0.003326792187238129,0.07575346427819955,0.01004944780818631,0.007789340129047027,0.2188695135251888,0.6940732534165956,0.1539079219564148,8.273090140352613e-07,4.435567715933691e-06,0.0002861385006073048,0.2004505626959373,0.08345589493610182,0.3249203322376549,0.7633275714582421,0.001217261834108175,0.4594608273733668,0.3653444084063794,0.2274988524155691,0.5449014864343116,0.2319659879292893,0.1453573346677839,0.4526267786497635,0.9553308424446226,0.4213301608438767,0.7104345790239882,0.4861690915359633,0.0008663528651364953,0.003308711181184889,0.0001412383694349414,0.378950655947527,9.429564960674394e-05,0.1950049912806182,0.04225394597533961,0.7504698150773303,0.002285156942181516,0.03590012841081633,0.661831793023653,8.349057926686864e-06,0.08697830701094943,0.3883501619885835,0.7265408771260601,0.03954616649671174,0.1320286113704765,0.938324350629306,0.04303751935340903,0.1065451024888522,0.01169850694883784,5.809471251061139e-05,0.184838821298558,0.4284072350068576,0.4813464387279623,0.004557373732651494,0.02232916470991189,5.027824574539285e-07,0.03599157590525117,0.609735449640545,0.8782749161721333,0.8755589837667178,0.9427193873873487,0.02514052172894063,0.01936625326604151,0.7902250619761442,0.003716868974278771,0.2125500839175402,0.4434406355664643,0.04352643785094961,0.2128699959540228,0.4722584366965388,0.03426940648842013,2.072706271725939e-27,0.9482380863937945,7.333076737371372e-05,1.549722129165928e-06,0.7910711369357196,0.1551414223938925,0.9051302704196501,0.002503047379960426,7.762507236282258e-05,0.9524919743482566,0.03901953880751155,0.4073651919160459,0.5022601725942661,0.01931978510828769,0.997749816523652,0.7544422931545985,0.2607372479111307,0.001636000439985307,0.3316920586165719,0.007698991621304729,0.1592484060193947,2.06798684481466e-05,7.214323132752849e-14,0.009790545741434724,0.8764409178947011,0.8760602917076729,0.4419521434325333,0.01921569153963925,0.3548140796479343,0.5857633913117014,0.06432567694839253,0.1281139060564646,0.544287737375672,0.007242174989327292,0.003312025875010481,0.00121179398328714,0.3904606910266455,6.512983230046555e-05,0.05918901236776648,0.1853411134664369,9.350784937290481e-06,0.09389945382503141,0.007852217855420235,0.4152727162369471,0.0463172920116274,0.7425590584777553,0.1302923224717469,1.281145180075509e-21,0.2271788971533603,0.4590763089654884,0.0003324259188200386,0.1902206862238024,0.03842347619195393,0.04031262553750017,0.2078992202427942,0.08859546280557644,0.421651573819744,0.01249392048204907,0.4761579165127808,0.5707838854182312,0.7087266231513016,0.5139746536968679,0.000303610468084789,0.004657252200279917,0.00652082410250973,0.2691479911661022,0.2282269055351131,0.2323323792656682,0.1631088579275342,0.4884290006774706,0.0761211723319145,0.02883749187204198,0.5644373007612211,0.0001247237008967752,0.7886515732669133,0.9937226776302859,0.000579214506972422,0.04694392939284727,0.8844118236867099,0.5941712496388377,0.3969849236445868,0.00605739234120362,0.9075904947207616,0.9886624277832283,0.08159512030117816,0.09004484768771565,0.0508606627630593,0.1235363646880921,0.5236994579638868,0.002005824651769215,0.01116756744018178,0.296014330476801,5.027824574539285e-07,0.8734121229811976,0.1349963775809281,0.04073207195975514,0.005801878602109888,0.2503328884234515,3.552294205602479e-08,0.6891316575068417,0.08600736996293964,0.5049214732390837,0.9796135184441174,0.5883954553435581,0.00109064902028138,0.8687614269233123,0.4218835610160609,0.04525058664819044,0.1975535826889772,0.9543405986142123,7.552836395024758e-05,0.8123019141033492,0.5291811815894597,0.2276711773571728,0.5300285111235087,0.003055970587376736,0.6408172828474565,5.872365770557808e-08,0.862282140384783,1.356119820166074e-07,0.8895988707032888,0.1731819733613915,0.03913469443828811,0.04673060329141841,0.03784135008280219,0.0754276967519875,0.6429379618829858,0.2549211165088527,0.01663812025936249,0.889998172715418,0.4327929161475383,0.1011488533219198,0.559743540424213,0.0007287749825514723,0.7798322169422667,0.9964906732910624,0.1665135912631326,0.9579682371170342,0.08471070692120948,0.113656684686888,3.019347518150332e-05,0.3035269271846336,0.9486180350791249,0.8553190384775331,0.8498847235088017,0.007371584989587996,0.001190115057364104,0.7071216716699456,0.5530121604076909,0.7181791891613497,1.887525339283553e-06,0.8144273385661703,0.463413946425463,0.004122909089768351,7.251775970602787e-07,0.92273907670411,0.9738873391687207,0.7335745536524571,0.01351250022139297,0.5960392313497541,0.001025406504377414,5.027824574539285e-07,1.148687550382355e-17,0.03642235588398079,1.399411078415019e-14,0.02082742628983834,0.03398007909537574,0.9900811214283742,0.2185555068723732,0.2541334389985817,0.9560343473144792,8.13283123505929e-05,0.325721466016468,0.03380629626723992,0.1646913381789313,0.7497017195672078,0.1425667192327002,0.09014638572122775,0.2817843078246214,0.06082130914179112,0.03290545577753242,0.5545832741677524,0.9667249776203444,3.63001573229023e-05,0.1266731838251142,0.001239692960175641,0.08875906962223332,0.001961398325616616,0.1378804866074365,2.775861462111826e-05,0.0001769709154649431,0.01410157009935544,2.922054775997834e-05,0.4098082331634268,0.909984737981573,0.8819077782213309,2.040634290195045e-05,0.05587354064361547,7.197189986218952e-08,0.8487628549431723,0.126958258769161,0.4722584366965388,0.4346256252842931,0.5380874690572496,0.6543088212089084,0.0001866256362824408,0.00752689822035851,0.8084403921553645,1.45379229428548e-07,0.7458927767801636,0.9371659701692615,0.6763610439148934,0.01222170675094309,0.6024856702234119,0.9976791228941412,0.3989365369411896,0.9112656104634721,0.001605795218436625,0.002559438842347276,9.008989412772003e-06,0.411703827508285,0.0004990696761062042,0.8231680191060285,0.2316427586443597,4.243705322984454e-06,0.007126936150419404,2.51385988425173e-05,0.3905542795803599,0.2541222073335381,0.002693580755493397,0.4040321162379268,0.003904919312136043,0.1485029122374681,0.3507022913270373,0.9427071499950915,0.1544193976038045,0.3853757231325587,0.004472086336167342,0.001661761198766134,0.02319749674940908,2.479465764950082e-07,0.4018721561627105,0.01541689436449198,1.621286516125355e-06,0.00642314812144799,0.0002446229272187924,0.8468710089568001,0.001272431946582872,0.0112619426786251,1.872177190525627e-08,0.8721230104180533,0.2125543183153146,0.02239865319818957,0.001246694372649445,0.3411807873613146,0.8387915840050847,0.001054946321496072,0.002076647329714691,0.4341388757066937,0.001204720622657692,0.1313271814897368,0.4736139789713975,0.7203617066932856,0.01297111666351387,4.127238041429685e-08,0.01381185268998539,0.4698821346324237,0.1350992679892284,0.000389404730972475,0.4903354269707863,0.003299412289664812,0.004336812607012824,0.8247932028284967,0.5382089184983057,2.909002215341145e-07,0.0003175225263339686,0.478055461633243,0.002642271940135668,0.89530380068149,0.02382200740215572,0.5017933212389952,0.4982971553090842,0.05772189934565101,0.0005229793532189474,8.032001700538973e-06,0.1932177875417304,0.08519847410878809,0.01578041042190493,0.02208426070926452,0.0009789206919223531,0.3648885995861608,0.001224093088288564,0.07748104894763669,0.5087640325179282,0.09077086077147732,0.001440079511683567,0.376668170310481,0.1824515272033647,0.08764310086358687,0.005786092665904972,0.0007111683262579152,0.4281175743074068,4.325529066366057e-10,1.102799777941532e-06,0.8276381395585667,0.6063812863854263,0.724828710036411,0.3914006404783486,0.02116511924161986,0.7417775491710438,0.7309861234970078,0.003740929059703379,0.09500664996232416,0.2559429530857154,0.001028685434085565,0.008316460197520413,0.1186733403937005,0.006321042104767585,0.594010862964874,6.629680674798981e-07,0.006127598673035493,7.313072088962828e-05,0.1098683506766158,0.08561158602140963,0.1486482043874783,0.301384887904671,0.1073110727364305,0.09490917649597665,0.07443583322443778,0.8417671936750322,0.002779451196249874,0.008715210608881039,0.6378611260065927,0.8294954916541089,6.355672466598317e-07,0.0001122872764314077,0.004698300932460023,0.1408344636840606,0.006249111330587902,0.6614459764256787,0.08257787728839401,0.0002886302635753539,0.5248808959194099,0.9935989078443038,0.3468599240522401,0.5830921038678565,0.139799472071477,0.005313900632920862,0.0002399044469785083,0.4850226591495749,0.4944617242417446,0.0003707563824047295,0.0002753221614524331,0.4293670909388911,0.5184662018336651,0.002186033505148913,0.0005146796603281997,0.006839072402094561,0.1912792301196026,0.8811348616346973,1.334171170561295e-08,0.6093363220427346,0.5373179685141054,0.7545158949457419,0.3019113562062395,0.002352814691286639,0.8834720138954748,4.2395927486238e-17,0.7066927253573404,0.000429157031920381,0.834681083077617,0.1081604576796593,0.01882509689663176,2.42869574250349e-05,0.2340222558961302,0.0002369097512529797,0.1799299572708256,0.4572774730832227,0.5005625542302686,0.7101373987821097,0.4288769326558183,0.0004624823023643293,0.001626782463352791,3.832413561558955e-05,0.0918784666757366,0.713220255235972,4.017009678331466e-05,0.3373246593141367,0.06527811915846052,0.3254203846082729,0.02451989538533117,1.215665485034221e-05,0.2529446634510084,0.4943921461732978,2.922054775997834e-05,0.1889447662316982,0.4829644952234267,0.003452418807334415,0.5683316198656433,0.01500446775207743,0.0153833304426392,0.01997718982996033,0.06274913416918292,0.5984244883332864,0.1091542281273583,0.001469703745213324,2.613003306062269e-06,0.0001334614783409311,0.04359151949709654,0.01198946877412517,0.2775595699496495,0.004718350640010798,0.6300427060591772,0.4507275199029139,0.08042515974539252,0.1813845615302615,0.032300709983556,0.6998230429998386,0.02096185018575619,0.9990723140281231,0.415730411211156,0.2885993544103613,0.6302001594036244,0.004249653039293176,0.582095480317011,3.871483194812097e-05,0.0088265200029943,9.258810918614884e-31,5.106192112564982e-33,0.2817713156910994,0.6284622700524307,0.8099388082353778,0.02367025975454437,0.5976864562354408,0.06905144787489829,0.09817432033028588,0.3351870013888739,0.9877909072459107,0.4507015566067142,0.7641975331731772,0.1159443239024476,0.980819406974376,0.9167416871882991,0.8282649515862307,0.9531497198175617,6.934420396726657e-08,0.1410731246482471,0.0007446716779373589,0.2303744305866311,0.07303287512947201,0.009307877836903221,0.0009915170163942008,0.2534869185766732,0.01087392844522109,0.1138426360780327,0.5165870938843206,0.8568862452067008,0.8873527101384303,0.2200743878671915,0.3685467416677955,0.02194451035168864,0.2204956534004528,0.8304129322816477,0.3488236767677803,0.1781870836927747,0.02787890868281619,0.2212131753774612,0.04950116517361722,0.03112387170563573,0.001417708759507125,0.0001333367854030521,0.002420835566323146,0.8783928109525093,0.891562178225777,0.3349946872801799,0.3245345406011998,0.1194077324275913,0.4171275489728488,0.1722071056130867,0.01781127104413418,0.4030880145760313,0.3904606910266455,0.0312883416575219,4.395736144574197e-05,0.5867468408225883,0.06209398080941929,0.04490607361517637,0.001012040745751496,0.9648598480122718,8.504755405412216e-05,0.01331036755033948,0.00605739234120362,0.1061162950092692,0.02949841658523903,1.726691461428792e-07,0.4141531585748275,0.2980717578780677,0.0520973282910973,0.249176736835189,6.151188078896365e-06,0.0008110059594535219,9.938972904731135e-07,0.7632865760377388,0.1411281365408646,0.1680734831816569,0.928639994257329,0.6586913983535965,0.6616660038237662,0.9015245125758632,0.0182546088465596,0.000328490357574269,0.8710783967963328,0.0003125850615302683,0.7166630783696201,0.2441367835960459,0.0009505346312610915,0.008336218294439453,0.06730714611629765,7.422888846580904e-06,0.1954310892736873,0.7799346922041229,0.0634136576276363,0.8951236248189153,0.005279974753901392,0.3338514502183282,0.002379675769618932,0.6628707849167981,0.4725012128467054,0.2125543183153146,0.4260516005251273,0.5213621005507816,0.1195880989364225,0.5715449933825525,0.01494155695175306,0.0005797508702575802,0.8441957509835005,0.003581266496618143,0.9879823618662967,0.1057451919623326,0.3329918147532934,0.09249757476310387,0.372638512922928,0.0001166313683451042,0.09173681340473129,0.177698550584659,0.4243696619766324,1.703954345903213e-08,2.176352169565918e-08,0.1175730113012047,0.03808837809795088,0.01182721737391254,0.0030046757816183,0.4122940004860052,0.1545266537593616,0.07948583397017117,0.8799442430489973,0.1189677363162178,0.2194453904220377,0.8849671031508964,0.6809094524139143,0.1212093966318925,0.5589062070817157,0.2601859492917939,0.2395648003504091,0.659518366238902,0.2517968353525634,0.282268904134888,0.3511062444832618,0.08016057952540057,0.08120616160539434,2.10617201010704e-06,0.8389126253324749,0.8015860619078314,4.003155444919005e-05,0.0002664623442678182,0.0001522776531777039,0.1449958601786839,0.0434789369355832,4.533177093030135e-16,0.001222796408661024,0.2596559126465215,0.5752734633092093,2.669688383589896e-07,1.752352244526379e-07,0.00982762068134614,0.0817071945216359,0.2489630094441129,0.7597926633324557,0.0247020157496858,0.6924809684482993,0.8023517781456845,0.5510495969831998,0.2406353100505265,0.01770684889770349,0.0003191594246711971,0.0002410343537733204,0.0003588253538224643,0.4377595381550423,0.5021980235554665,0.7075765352305143,0.4411307403649212,0.6555437341014119,0.002470252302739201,0.02748304150917011,0.006900242125443201,0.8812962901608981,0.4922984419426187,0.6409598927310841,0.4434228728138658,0.01616177400169869,0.001626288732832932,0.9579682371170342,3.900546009818494e-05,0.4670706616653446,0.2452578982300722,0.1651583162822035,0.967171793221364,0.4901095158427116,0.0002975090591319996,0.1044014605298139,0.01616177400169869,0.03197377934306721,1.468399654191209e-05,0.8010052100483689,0.0002223399085273033,0.2194453904220377,5.102576265009005e-05,0.1350348510442438,0.01627034705193395,9.041665489360211e-08,0.6167427945919683,0.6777390035352915,0.2857259622774401,0.03062165772889796,0.4017518800946802,0.719760842653777,1.007595161380229e-07,0.01541689436449198,0.6930547513082762,0.0009291992698566405,0.3368240787394464,0.957214834460033,0.01943370717453092,0.3411727283693062,0.0001889433313776612,0.1374290436195703,0.1149304487612594,1.411651356293797e-06,0.02567753868464094,0.838281589565302,0.007772172429123348,0.2483689070288658,0.05080532731901217,0.779835672000411,0.7965888150972729,0.4024144441408408,0.5624278832261533,0.0022123604539981,0.0002045223517004014,0.001675629381076889,0.2133673363746107,0.6009559471850662,0.1583430845521221,2.997788172959974e-07,0.6433574699205923,0.002516962399772362,0.3276158663941455,0.685339599623771,0.001226687812854325,0.7454003169984916,0.8309675909325682,0.03822749291262847,0.01143551761738947,0.124726721831177,0.04591733692658292,0.6408362028484291,0.6095404457380045,0.4838594388697957,0.01249234425973277,0.2146079377100721,0.7689714695996296,0.1171293765217985,0.02382405450472588,0.01027305231114932,0.2212947605289362,0.003358114110926516,0.4466033965311679,0.03091420678328313,0.007818930478055723,0.5753415958867029,0.8005529239463015,0.1311539385658451,0.04124828081703938,0.1088655460931745,0.0001062930160236758,0.2078511628859993,8.050995480751002e-09,0.6891918561222889,0.001224093088288564,0.01173117861014916,0.0002988889585799151,0.7214146480759099,8.460399739216017e-06,0.06960086991494752,0.8867961316409385,0.8898744786279353,0.003130193616847407,0.00439635083896721,0.2058619251683906,0.5276140506738625,0.05094624694006621,0.1239567234380103,1.700693096265803e-07,0.004104633744265633,0.3409572550684294,0.08677779250340041,0.6892431786874353,0.4055055713863726,0.06246995038746442,0.001249261981009724,0.001301078460233511,0.9799604837772428,0.02878588142437173,0.3588322316028004,0.0007975821042703478,0.0701151764061308,0.07237632203303607,0.7398698243078285,0.184704262530644,0.3743048770390933,0.09034424937609385,0.06082130914179112,0.0006185746082269518,0.03649930875822276,0.0727922485482585,0.7323860466692993,0.3761062153511685,0.55313648042341,0.2414091531061868,0.8493422468773305,0.01267224409612773,0.01762922340348568,0.0004282721955786109,3.619905814196163e-05,5.076062247465681e-05,0.04119680756027107,6.137364583577376e-05,0.2705094098917692,0.0001763116228222014,0.1505740046780516,0.4040814406919541,0.09882929442604402,0.0009888981125452912,0.882676011695239,0.1089670374667021,0.396895667015494,0.00052031596641664,0.0001007523813841891,3.083075055534709e-07,0.3615940224569189,0.04609426597947915,8.71032455694319e-06,3.92534084274675e-14,0.08710449519531127,0.006770124778891353,0.7056041206367637,0.5918869301186455,0.07496613678501267,0.03574088299518607,0.005791653066188022,0.02005629908575101,0.5546732744918126,0.6405766724402243,0.05446316778449708,0.1156463936833632,0.4579456916050176,0.005471522217641137,0.1002473659116109,0.2167014549163011,0.02089542168853643,0.0005822530582049018,0.8311551074480885,0.001267757923809842,0.1485029122374681,0.05268044449105929,0.2402952953718569,0.5914346361891176,0.6672154138382111,1.251847273728666e-07,1.448855066834915e-08,5.239239413689494e-05,1.472457450157161e-06,0.4897981867280332,0.3870029617881414,0.1883424115321229,0.00684893727543246,0.04062431569382082,0.5403022032234097,0.1080494894779778,2.05217364909851e-05,0.817064173171643,0.6804202073420909,0.9527738886619946,0.5210963633468754,0.05962277406479295,0.4186605353597023,0.03831646449068804,0.2133673363746107,0.001366586869502636,0.008851453532357495,0.0001313649950374314,0.01650905741270045,0.2537840874284416,0.5510064078498074,1.648190597919355e-05,0.01249234425973277,0.0004571620160776645,0.1908461129314542,0.001216006121292186,0.7600726403364612,0.02338317314516361,0.8127062467090734,3.604024048413041e-05,0.05822860744429167,0.04207591005251504,0.6447659665917317,0.3351956167064761,0.2633279341769861,0.2523050870894342,0.01240880994299362,0.006220895673199392,0.0003808156920452564,0.1393411797487379,0.4722584366965388,0.3135290282756244,0.9060020163937786,0.2809292279087133,0.08512768724742253,3.471579566862568e-05,0.0009237680583671254,0.2028986497674972,0.5097090370587978,0.02282423711570113,0.03207596172209569,0.0002343482239837526,0.8143827449048033,0.01312041588612147,0.009890489819050838,0.8350611207932529,0.008609736693208929,0.02051555650873487,0.9590397643373596,0.1575381352960901,0.8381705512621298,0.2185638505273189,0.0002955435366051787,0.01798873836244909,1.491950764356809e-06,8.647490972316573e-06,5.873814010388978e-07,0.07505439098056518,0.1175360664271995,0.02546164384265135,0.002983180553213911,0.0004679812773117365,0.001284313999734505,0.0002324657924152045,0.001103111617383244,0.3167064326926355,0.2293247388566408,0.1197898655709426,0.519132942494467,0.06800713231678443,0.3019519849494733,0.03815504653187678,0.0008345001995544057,0.6757999602875142,6.74849474219571e-06,0.9887396605254718,0.3279222570437829,0.9078770692133724,0.1946154285440945,0.0007949787458741496,0.0896953974437726,0.009335007772597778,0.8764944342949155,4.921380102350873e-28,0.06214100670966974,0.06912362795252523,0.001246694372649445,0.1084256050669293,0.5713361082212909,0.1017965608620761,0.9518323344757121,0.3770662306876115,0.03575829217217893,0.4233682115645848,0.2193316253953126,0.4651164434102207,0.00574537439870312,0.7093246589491564,0.05285171594916185,0.7913946718773649,0.04223781989187476,0.002419614730342473,0.09456025904973937,0.001549967171559093,0.06619466489952879,6.024787754729966e-07,0.1317918429111272,0.221739064260269,0.7570514425346156,0.05253084117115773,1.313956810740554e-18,0.03776930872751095,0.4260271418057532,0.0003066061133703597,0.2078355400291711,0.2767368635144892,1.346792051168899e-09,0.2531324903369526,2.182525997163435e-07,0.02403331481153059,0.801707004782642,0.04359151949709654,0.2714180089809339,0.8415730103256502,0.6781899423048977,0.06154507843898502,0.1188554049651782,0.4562599421848899,7.72253309121884e-12,0.7087266231513016,0.0106611259355938,0.4803110257813204,0.09645469837649613,0.8843249868668023,0.08859546280557644,0.9192185515204666,0.3036061308389687,0.006563583009027573,0.51942114090628,0.3381982189631553,0.4098772185940674,0.967160322054779,0.4022677457945175,0.2189237118108892,0.1777188699448592,0.1755218626516055,0.2381774400523523,0.1714211890628156,0.922861263378296,0.02225264588105353,0.2992963849370398,0.2223125055236712,0.002473683484392214,2.534839707901582e-06,0.005777123126312498,0.1241384205479276,0.3495380893895135,0.4318984007727337,0.0007761516373113436,5.22171854681329e-05,0.4906792188667913,0.2613347711136881,0.2595164919154243,0.4220067851084847,0.006629926828404176,0.5746519284957546,0.1059126442774592,0.3881030807632245,0.2144591514836744,0.4470816184506266,0.3799225006226438,0.9407974950971635,0.09230973168228518,0.4657479678363063,0.6777222383531695,0.2776994531900832,0.1163798369001831,0.6315380226558112,0.1997909207306224,0.334829113591316,0.9799604837772428,0.001281548678590978,0.0003136150127533189,0.9362235521451817,0.04871345413530737,0.3293655392152437,0.006270307565059485,0.0002548704472947262,0.02651241338889049,0.6620711622399017,0.1762733520817116,0.06214627216967256,1.522656312110304e-05,0.200183075139364,0.9510177664294567,0.003027831736349833,0.04050446909831432,0.09239976654766829,0.001492474409697261,0.0002369097512529797,0.08420916993047761,0.9396357150166947,0.02751664539961787,0.6085069557650185,0.5227480742840499,0.8622599070722872,0.7144291317971496,0.001301856725794189,0.0002782934745484276,2.371231817572846e-05,0.001193160181890549,0.03398007909537574,0.0003721945486218658,0.04124055277487652,0.002883615889497239,0.4645245506402589,0.01574429713248588,0.6872446874667441,0.5283644472740511,0.004088824090026716,0.01096582189198587,0.0001436723444113225,0.04066989833489263,0.09247833666265665,0.5695490711513914,0.01990356600270842,0.0001914613656479616,0.05430429418721022,0.2537366611744146,0.002345061583879018,0.0002711783733218243,0.001091162580844866,0.01957644575272414,0.601736551744219,0.6421127216926329,0.7458047097891749,0.4652876839435759,0.008020015974935856,0.001459746366656261,0.1038629946199084,0.2202281715690612,0.2006182006366401,0.1321675043660592,0.001524116776893743,0.560530894986495,0.0001911348842768411,0.01675102337728681,0.1181137567634173,0.9908278124918487,0.000394986016396553,0.8755589837667178,3.344857482458638e-12,0.2566366774748292,0.08908573316357292,0.1739115894593544,0.006812368327992534,0.5543363277401123,0.6383102132781001,0.8889024906513561,0.02199195275006046,0.4973964033983564,0.3834446433022361,0.02319668378233647,0.9746071238869031,0.8403984336163046,1.242784597672283e-14,0.09605631294586436,0.7975091294346375,0.0002434373260347569,0.9733823414685572,0.4245644849051275,0.5114149891880676,0.3649960617228145,0.04196916008709328,0.9337651105246443,0.2234210866577817,0.3658543575251947,0.08042515974539252,0.01865685083313045,0.1415617646504425,0.7366778609783335,0.6476769332668106,0.0001835014729351654,0.8768267593307794,0.7871448737116492,0.3848333277545393,0.9855865427273619,0.7477801802972881,1.773874619853839e-05,0.007259452762367574,0.2369831964884495,0.7624402769476776,0.2143750550420698,0.000354633355023671,0.3912480362348831,0.000339197733692133,0.04018393332025132,0.05563901322792007,0.01735686767191923,0.1286639662852072,0.8467230836712114,3.314967819270614e-07,0.5630513185045434,0.04406916379453987,0.004267949779920168,2.569124885656152e-05,0.140451890548283,0.0004652423750396078,0.9002960875310292,0.002552954675713768,0.207747362860353,0.2577145436742577,0.02445382970670921,4.083255446205262e-05,0.8122777077752956,1.857694029548877e-09,0.001759592660570554,0.09650516904306981,0.9953662136556667,0.558089873567818,0.03232920636492624,0.1808310898366843,0.03371224703483799,0.3034540027992411,0.8599029043656278,0.08770541026379636,0.4253596427506191,0.0003408404464409555,1.16055499343865e-06,1.919595767552642e-06,0.301422981300879,0.50346629193641,0.4312723312179567,0.0001390928009328749,0.2147545105636198,0.2241284391345537,0.0354736409104148,0.2858013268351996,0.003283854897631433,0.6341313402416529,0.0008600208314980929,0.05796928138849274,0.8252332630504889,0.9590397643373596,7.088330351726355e-13,1.285408171202884e-19,0.5575565091789768,0.6068893579166885,0.7487421736532451,4.501661270669388e-07,0.0608434011939895,0.02919349944358048,0.01173117861014916,0.3655393200830152,0.835354428458382,0.9188364795036531,0.3520537376507619,0.4821268714381907,0.7535660221128655,0.3972814790836413,0.7456781815562178,0.02422989031596563,0.735423134191913,0.01705534368933105,0.09817658262725368,0.1581829948254229,0.3228037764467038,0.00136175627320675,0.9807571904188169,0.1934325311037006,0.001858498598473736,0.0001718658851900953,0.07382731932238128,0.001808932125036554,6.009618402147859e-06,0.007161176594776164,0.6980657127504927,0.4043245167505359,0.2225569449213017,0.000480985911688747,0.002122982987293108,0.006147592162562712,0.002256792471410543,0.5984343495597917,1.179535836783614e-06,0.9210329328674836,2.552310874006704e-05,1.157795376282835e-09,0.002192934390604757,0.80318359661627,4.871370408056939e-07,0.6658306005252826,0.009819447773174365,1.324567610980339e-05,0.009890489819050838,0.9680025018692554,7.200105893716193e-05,0.838281589565302,0.9954203167841977,0.001267757923809842,0.008157405668029317,0.2261327747338988,0.0003738408782297425,0.5601434880450308,0.3019519849494733,0.8469562149725246,0.8286991588516376,0.1398228499987772,0.8511593722080151,0.543124666785875,0.9693140326527426,0.2076223058334166,0.5332925584845176,0.4739623955792112,0.0007034890578855505,0.0006771129189422717,0.0853524835626169,0.00524080462880584,0.5396109859119631,0.184535677432065,0.05248862042662705,0.2496338224404132,6.466671517670043e-07,0.7720888135500441,0.3663255015012436,0.003547439969678624,0.9544308260931004,0.7446221204799977,0.08972463673135223,0.6420904559918,1.966849242003586e-08,0.0001252240499629114,0.0002090942968535982,0.0358467550475823,0.01372702976967304,0.1091956174004123,0.9931593647709804,0.2450070332911796,0.2011705623051645,0.9410630604870223,0.2053172930797812,0.6117801829462806,0.02801176932850961,0.5707838854182312,0.3681905288851504,0.4702987995990847,0.1000298811927505,0.08956502303070284,3.592364328287623e-08,0.000583807676683604,0.1094012654434308,0.6641260343158324,0.9319033734380527,6.087899133143676e-06,0.6787534087382657,7.538263746695122e-11,7.755815645098904e-08,0.0916638022803658,0.9488068977974551,0.0176909400794097,6.199264473071528e-06,0.0007369501974678598,0.5853957555616968,8.974967791249523e-06,0.826992147773028,0.6038908631915254,0.001025199774137296,0.8844118236867099,0.004384534722658638,0.2700517433571867,0.4298826909625206,0.9078770692133724,0.003326455524481337,0.4260064784802361,0.0001068310674208404,0.008025463731810496,4.298219622835031e-06,0.6792488152227492,0.07394323168514946,0.2986971370738854,0.5392649870733451,0.03307558890213039,7.927159128270884e-06,0.7785679100010667,0.1044014605298139,0.7259012974530684,0.05995189959557582,0.7271815456095324,3.37727358489923e-05,0.1704681447206409,0.07097043872331162,0.0469569207283024,0.7477975656594543,0.02831988142392382,0.0103744052131774,0.3167273105795051,0.9577315007099367,0.4711763672562978,0.03353857560997232,0.5578531456439428,0.4560333336207188,0.8118341667340282,1.091595583965665e-05,0.0001252240499629114,0.9482380863937945,0.5909229653767979,0.0002119547392657003,0.01372316090322501,0.03742003636739624,0.1332391245890643,0.6855937234634061,0.009595048933210892,0.954938950227045,0.9534553094741761,0.3806907730961141,0.06603401734008117,0.0007394510755493657,0.6698901210493172,0.4943561980797452,0.0009727021657318745,0.958434954909163,0.03410053507473688,4.372902987796047e-05,0.0636536473863465,0.5838104091168859,0.9939109395728927,7.300590077753151e-06,1.660937283573323e-08,0.0002433804677414921,8.813937324880945e-06,0.3602173173815276,0.7398030222933282,0.1118076927304302,0.7801583390298356,0.02199557527633737,0.002529510655886249,0.5815987925205748,0.0003213889032017145,0.9060020163937786,0.0006625190077894625,0.4407866405677648,0.0006189788726166221,0.3249188829337193,0.9455345234220723,0.00346034319326849,0.5578531456439428,0.03953317179080033,0.3650529857395265,0.1602621605539953,0.005073708943730734,0.08168548994190418,0.1239977259109246,0.1277104270370585,0.6355051607076323,0.7013678809178121,0.8690405171724176,0.0003177899904406463,0.6169686680500346,0.07172514608669422,0.02083356474132938,0.5061021303641522,0.9449580636486389,1.098671649535689e-05,0.4144754537606743,0.000953155086623929,0.0002158552226804994,0.05657378567654567,8.469190791960077e-06,0.9282757403679216,0.249291449248891,0.2626737613994291,0.967171793221364,0.0007640334369660013,0.002085695387277933,0.0001974586389518773,0.03867740963207311,0.9060020163937786,7.599554792586779e-06,0.2655766413698205,2.115076012556295e-05,0.4180348995974108,0.8111157792694961,0.00569084459921002,0.1646734409239844,0.03864536545140895,0.7899509109154192,0.2438366008161,0.8373722981890648,0.2496323178383083,0.1236348428990091,0.4880311325945096,0.4595696897001346,6.669773663211107e-06,0.42032755857978,0.5412772094620463,0.4132460852655823,0.02584755766550295,0.001464849379704587,0.9034137997304608,8.338100935111098e-06,0.2728507653536406,0.6428647840094251,0.6215865309004218,0.6926157488206697,0.0004781792143631867,0.0009347036183554978,0.2182891438810337,0.07017683861413626,0.916178359361523,7.007619150155685e-05,3.774803997640027e-24,2.551575354676043e-27,0.4751428851829839,0.1833332834149041,0.03927381506696281,0.1137305067539267,0.001335250328797658,0.0003143979839983803,0.001148808467676792,0.7765410593665456,0.6479207417887568,4.073862150915494e-05,0.002538432530777004,0.9549053012033992,0.6061123567389116,0.02859097390005865,0.7453958782589798,0.177668161169503,1.205462421177114e-06,0.01532443849377301,4.232870951804536e-06,0.0003125772012512559,0.4513446260658638,0.9140495083200286,0.8218276621854582,0.891893336614878,0.01871467200854308,0.004498742990159113,0.8946744598715128,0.5594465031573188,0.1349963775809281,0.0593343382042598,3.149512082996992e-10,0.9248702188000671,0.6523300869301871,8.713824928186247e-06,3.455044032518921e-06,0.05622768469245878,0.005158168482350563,2.143229274990709e-06,6.692887049142194e-08,0.672898626361069,0.1221284705791252,0.4821268714381907,8.506648192580555e-05,0.0002039609842323556,0.9639058533246051,0.0952458574418225,0.7745327958319056,0.6905927436235108,0.625339412067657,0.03929298850169751,0.04518048135181654,0.3072668490671006,0.02470605055062047,0.1922075088534126,0.8462213423121087,0.02647354791114953,0.4690411666352498,0.3165951065323092,1.772410256119997e-05,0.00767504250311159,0.003224081434452812,0.4366419923120466,0.02768270339164583,0.6524212104156987,0.9894953227737648,0.6701635421875277,0.8460126507081173,0.001761440198068535,0.4254939561134368,0.6445819403425512,9.214151289025983e-05,0.9674435008362836,2.646377699496787e-09,4.632949063595431e-06,0.7143778801135354,0.3876929978116246,0.06352747372839954,0.516593740540374,0.5018307597170115,0.0007201351723365604,0.3880517692122914,0.07280137347965625,0.000365564619262693,0.03757395891229506,0.7650954396153811,0.07304052817791673,0.01862259582521062,0.002856408678450628,0.1507418737821729,0.001532682631762309,0.2534871979738717,0.006163503895356845,0.00892032457877886,0.8543889225083733,0.1197680194843393,0.01798354559578303,0.09913037331005423,0.04238026424311674,0.1739115894593544,0.7481791870301072,0.00183596112569291,0.5399776431496881,0.004945644766594188,0.02286834376243846,0.02242955281345583,0.7101373987821097,0.4142440935275727,0.3054080815444912,0.01029199662270448,0.2431999758895698,0.2510312873325661,0.3982221989131544,0.0387458590316813,0.406505234165221,0.002953628948268121,0.1997584527136076,0.6578620898078757,0.0007977189934625198,0.416086859208098,0.1126439604381899,0.006037474031844253,0.006690004951218718,0.3233044706229065,0.8894497249893139,0.7372109020486741,0.3909535354000627,0.9974146874330095,0.8703359045074875,0.7248300199258522,0.3240815256220129,0.2311188486233637,0.001011012063592681,0.8945839571098519,0.05978089563452722,5.137098271324727e-06,0.6906568482841626,0.7526547842424191,0.0001893777310630445,0.09313735972188794,0.5379975116639537,0.02344633588801879,9.340514126845577e-06,0.8632157586879274,0.07068805796690851,0.8759581751942805,2.969026650734634e-06,0.1336744230933964,0.114698287861638,0.903486632302863,0.798248481118935,5.459922129550614e-05,0.6649787284657631,0.5559044883827835,0.2211568313086055,0.8680241263615879,0.2443175693611611,0.7658401433439225,6.905227853433574e-11,0.0493688740921162,0.002426132076399412,0.0002698863273626864,0.000137558014595522,0.3552051143418468,0.0001461101763021358,0.1974081347975585,0.009873612838993701,0.3101998396535777,0.3336930391692783,0.1763567036358286,0.02571712684365581,0.9952594379297782,0.8332937900089283,9.499170295052388e-07,2.169978395886743e-05,0.9233161040252401,0.5004691399282218,5.535545665826859e-21,0.0009259799889876023,0.9335390725685782,0.7214414435657972,0.244307904145228,0.834681083077617,0.02449523114892803,0.9068224432698642,0.9031598553555675,0.03158156157180215,0.02629739535217748,0.2421178908858981,0.4003591155312017,0.6203554101174856,0.1102416384616799,0.02926407029680012,0.9936399263409665,0.009530266258106374,0.8854536865078974,0.000192654728580591,0.38644517542041,0.005776204376819817,0.1841435494257989,0.01188192788102224,0.0007977755442747913,4.040958488012496e-06,0.002099856304975275,0.1233231432728682,0.01075791112872195,0.0008889243307466199,0.1403512825599222,0.5195091806704866,0.002243969310217527,0.800000982304538,4.841940147921731e-05,0.8528618293882868,2.676222646927457e-05,0.03802528424440524,0.9558826733176533,7.492530968354686e-06,0.0584502377581159,0.321061385613174,0.2675832819031464,0.5080039558175713,0.1692156673582655,5.131828255824708e-07,0.002549497935029033,0.8067752574346339,1.939310580039976e-07,0.0003996176947793556,0.0001166574815739313,0.1966425598466472,0.140919501305056,0.2711580328389318,0.001800568165888119,0.3106517767954498,0.632796948574615,0.879268547154315,0.9360378062677456,0.01227659316365955,0.004435959486785619,0.6768417726332125,0.99741029642608,0.2980376866977963,0.01019165737065085,0.1859703242323357,0.7115410127782014,0.2135537184498935,0.05791572900975985,0.4554315775288778,0.0003140589327308615,0.02917706343099743,1.551599642266645e-14,0.001235564203163701,0.001392206122806271,0.9117619935249506,0.3281743198620456,0.004743550717796171,0.0685823009061293,0.8071056643528347,0.005265217655995512,0.8867539594136896,2.815196112801652e-16,0.06009127515590917,0.001819815254885144,0.4726769481391523,0.006220895673199392,0.001298689214004168,0.007531816919219041,2.06798684481466e-05,0.7633275714582421,0.1175360664271995,7.68533741160415e-05,0.1994188552071836,0.08366292047190392,0.0001289388207721462,0.9964906732910624,0.00030952951611909,0.4851666739502515,0.9932069593053779,0.8903742013730466,0.338656482729815,0.7639663972490019,0.6921135402542055,0.8609507870379646,4.840242162657155e-06,0.1186733403937005,0.2202518991600624,0.6929504880479856,0.3776244937434788,0.02409333599248446,0.0003642842858809474,0.02778542628651618,0.001299704317269473,3.313546721693963e-05,0.009530266258106374,0.03406292353441732,0.7141059354118009,0.003828318132032211,2.448838209189619e-06,0.4184275365273842,0.01502412229299748,0.03200938571720034,0.8994178043026303,0.0008569514650500358,0.1195646304860424,0.327099332061344,0.1744766583382204,4.609910038062942e-05,0.003733663772937746,0.4668622003894419,0.009220858767888834,0.9487314723178432,1.480197387740987e-06,0.5907051723460404,0.9488235731874088,0.6128678725729048,0.1406515510108132,0.002516830434597689,0.005791653066188022,0.001514856745830107,0.3280036639324195,0.4216179731223939,0.07008264728834956,0.2404743314234409,0.130264128420166,0.697038916662684,0.6147554308037275,0.1366763379763417,0.008759121889553991,0.06226530202665549,0.002137343726343265,0.05744788213448248,0.269666958015048,0.3751758161675854,0.0288062771988102,0.6238799811745201,0.0314914006441479,0.0517457866153256,0.2605060704491454,0.0005700080568804269,0.002940505230966452,0.003481562434848152,0.4199169887323922,0.6355105617908886,0.002896478505682477,7.72253309121884e-12,0.0001021796389428214,0.936343067743138,0.001605795218436625,0.1031000156051312,6.214669613966091e-06,0.0655749433822459,1.166964897012626e-09,1.401494383167596e-05,0.1027372763767406,0.4554315775288778,0.121924778583823,0.002041776823972255,0.6166684993185592,0.6821793180102554,0.6002074398336865,0.06916573383298519,0.3488614770677054,0.8489760983102637,0.2674559165689946,6.969812241490599e-05,0.7098030582202475,0.974801628386923,0.8460126507081173,0.6543623219950002,0.01306730484842443,0.7720888135500441,0.5896926863515476,0.0223786906848242,0.8481206873841814,0.9875414512444294,0.0003218421980763774,0.22497442123457,0.7333059055364348,0.03500335780443945,9.963363580499233e-05,0.2558418503082229,0.9901999461317573,0.2231134982955826,0.7946670231009676,0.2953856886417403,0.8805754520012028,0.002351146348208757,0.4228808175119253,0.02896322605969104,0.6789141685473864,0.0002131786605031836,0.4203577889883934,0.5376825376464153,0.006843825048361008,0.05967213099560646,0.1142561768309885,0.9746071238869031,0.3285189098394271,0.3041732582767491,3.135736586435923e-20,0.0002868120728583836,0.2121228496121705,0.8513414482102312,0.9746384985077886,0.1747670983359775,0.747708359437032,2.46991336370859e-05,0.01810308784750775,0.0008661026878889666,0.886485069734975,0.2097472597936847,0.006136379463121174,0.01255306797702727,6.229774128528933e-07,0.09167731599421838,0.873610100510814,3.326219211492944e-10,0.138949276297292,0.9093005082696102,0.0002500458321304916,1.367212969477833e-09,0.05477624286598056,0.06293630958704677,0.2433375769443425,0.0448782450851486,0.04591395021018074,0.4588291776078409,0.0002195248537119524,0.1292661220062496,2.804658251909305e-08,0.1360629914994124,0.0001348910688780913,0.004057909245011869,0.8636916187733974,0.2337659169791104,0.0008718267272917907,0.08270315121838746,0.5091199361905344,0.8007497714694602,0.8747267953631255,0.1196252622827404,0.7023330881420468,0.2421178908858981,0.4632876869002128,0.1117463329133245,0.4345573604486307,0.01624982421332856,0.006751157520839104,0.3134495878066694,0.002273752316986457,0.2080727240190052,0.05030597856296501,0.06628611994227065,0.85409529474163,0.2876725766932239,0.001116045500781852,0.0002417929051842864,0.632449917674561,0.7294009399995718,0.04642433448792391,0.01578856228781265,0.4482137150858864,0.3671025849389755,0.0005988052972024663,0.006782940638672357,0.2078511628859993,2.364550693110051e-06,0.08661545834258308,0.5592455683449866,0.8764409178947011,0.6440908326383565,0.5158646513632711,0.1935204398333558,0.2007879179679022,0.01794720882255028,0.4003591155312017,0.8309675909325682,0.01507888285984342,4.177326990592351e-05,4.181384940195882e-07,0.661962502128824,0.03664684700930487,0.3347908322928776,0.7295897138823045,0.3872981113659355,0.1577498932810305,0.001617149153313026,0.0009640914284003083,0.8300310883815415,0.7805666358072886,0.0004266446135163736,0.8258845043219301,0.6929982907774326,0.1428294219047669,0.003052657659849219,0.1000236070832623,0.1505740046780516,0.3190483327614526,8.363150311848658e-05,0.001451496541032309,0.0277905383954766,0.0003711129156308601,0.006232342103266318,0.7542873151034758,0.2781361130053692,0.000481238902656147,0.4868785971992027,8.558021940009484e-10,0.8439947762371545,0.004104289584840495,0.00935043273330481,0.8799514093321543,0.6926157488206697,0.005185618185112945,0.03951273042806219,0.0183516726767906,0.8955006654104856,0.0001232239663652356,0.005263927522849338,0.0846836310709473,0.4563384861997958,9.467363265509422e-07,0.06211525059245335,0.9390323771213417,3.114958196995803e-05,0.02673544491260007,0.01894159657003108,2.939507772884589e-06,0.01050256224144307,0.001061543291330408,0.5052662152273911,0.690552639611366,0.08202752483422655,9.961529087342714e-07,0.001779494796082234,0.9402737741323599,0.2656628320208816,0.07710679457885418,0.2584598528516442,0.3477456169045572,0.002142215395544074,0.3332533381388386,0.7745549618640901,0.5774576583489239,0.6904459626049985,0.9021203131141343,0.01883858236848391,0.2894725091430159,0.1123176685575903,0.4228754051049125,0.2486909305190843,0.585407149949422,0.04093324585400161,0.5828391844729111,0.000462653228467349,0.0385669690210072,0.001084100588069485,0.89530380068149,0.004014574254198456,0.5726163610311308,0.009871976147165703,0.2459293647610301,0.8139489752313368,0.5728484855246055,0.1943556776471635,0.03372099517428041,0.6696499718500456,0.006169786997610982,0.2004505626959373,0.5183518777566806,0.02445382970670921,0.8589355940303313,2.557705318729575e-07,0.1145088843908671,0.2174418073082647,0.7810475804891237,0.003107797113722167,0.7353453742966264,0.2474932975299636,0.05916500089095462,0.9882269857857361,0.006827124792691808,0.2753838738268414,0.9581309797509039,0.5903845730073948,0.4243107465585528,0.09576318023199985,0.5491984016426618,0.8712657199642523,0.6528295936850514,0.07411409998816382,0.07862043938566696,0.765730458141459,0.0002446229272187924,4.998699278639709e-11,0.5527119465171121,0.5906460159447739,0.3735277631427608,0.009633191806127459,0.0581935916086722,0.3833844370752425,0.08677779250340041,0.01532789760293378,7.995022210112182e-08,0.7556201944954749,0.04629651459465477,0.2899356964264908,5.899550720580761e-08,0.1995264856857459,0.9188364795036531,0.7898399195306983,0.01219321006086563,0.01372070163960024,0.6608523495201036,0.2565138783464332,0.006328718023225642,0.5509976768121981,0.7687443901375322,0.03919141631845614,0.05009371472369609,0.01936478421687373,0.4245507988504668,0.2632082190657489,0.2847353085142406,0.4908757367707898,0.1694281683580456,0.01932626911298031,0.7532240459234625,5.952702638118534e-06,0.001217306295234513,0.4725012128467054,0.004921855491449523,0.0230912094530018,0.002982048765603432,0.124726721831177,0.4259422964890617,0.01856576469564138,0.6531736831458533,0.5196977811776369,0.7609494344372136,0.001384804873150879,0.09552530353029026,0.1937954708161001,0.6389731408085969,0.199554498571902,0.179971499137757,0.3316920586165719,0.002017313392433339,0.7686772093745901,0.122996442298211,0.9353451798448758,0.02598800115529657,0.06124204261892428,0.275881051119729,0.262134788389773,1.706912292142524e-05,0.1214997558508489,0.006973511712936865,0.5079950890958392,0.04258073868132293,0.0001550709355886456,0.3305699220150711,0.2039021323449248,1.453876969907563e-09,0.8878679678891161,0.24659675770847,0.07493088620364295,0.5037205149999638,0.8803315340772719,0.1214325147721723,0.4560333336207188,0.03068075630892725,0.001346184184465349,0.1791136343431243,0.5392649870733451,0.8740856582343864,0.03852520295079676,0.08441058029156029,0.02063275713008917,0.08831878258993352,0.002476241544372525,0.6088105494803008,0.1997182686836669,0.7158221957997314,0.03161725237881035,1.731976574528813e-25,0.1685699313478543,0.3878345078803444,0.003055970587376736,0.4242863564631523,0.7194545149350857,0.3517035730821587,0.03465550139895995,0.09597440790296674,0.0005149548194794965,0.001759592660570554,0.8898142649496492,0.01357441566595315,0.3116915650287649,0.8952609405147751,0.6236691779716369,0.000403483265489778,0.08600736996293964,0.4694909131082061,0.001894828650730304,0.4873049990213263,0.7797782609077046,0.09326931546747171,0.0003143084120434265,2.176432727129026e-05,0.7232104218827513,1.649817215257176e-07,0.253416793746251,0.5479800099466985,0.4710400190176429,0.003750700770603787,0.01250082135308388,0.4516190934714815,0.8116946052600046,0.009444674182333903,0.03074600879103819,0.2071627116400303,0.6311310220576656,0.4317164117309523,0.891131492131317,0.3011209215210867,0.1251755709966701,0.0661873099664901,3.010996291944371e-06,0.0003431146312460966,0.5809227485139705,0.3850223048882026,0.9390323771213417,0.6370890275775848,5.223221147040451e-14,0.009017410264120193,0.003813256135796502,0.04434092310910814,2.730921022267978e-05,0.05874337076464949,0.7490328385849714,0.0002965875259547349,0.06818026995287874,0.092941476480007,0.08810755349519543,1.961802745068183e-07,0.1571979754512086,0.00304578516407232,0.01936478421687373,0.4123639719407735,0.00368153431052856,0.9199540683529321,0.6681761326770439,0.1074198800523367,0.9103592600636222,0.2522614558746137,0.216053202847041,0.5332216180785145,0.5291625945880779,0.426180930695923,0.4238630865536168,0.8955006654104856,0.1680734831816569,0.8834770636725048,0.02517979732930081,0.009851361761945551,0.9438366638202217,0.02642388609369246,0.1562070678712724,0.9913671482362709,0.8519735615983446,0.1640442254764871,0.6906568482841626,0.000171195105344482,0.1823067870126324,0.09317971798810486,0.1521286851613627,0.0568965244907855,0.2226445780796723,0.3301134079802281,0.9680025018692554,7.829731487158564e-06,0.06584883157539072,0.1013718710992879,0.1141567309066467,2.793004240195247e-05,0.4345573604486307,0.106294795629223,0.1065345052007851,0.09160235702808897,0.09136251615333399,0.5332945480625294,0.08589938362499987,0.02281972918809521,9.015096021913711e-05,0.2133673363746107,0.4914775420420692,0.8015860619078314,0.004991275765866232,0.02805650240244557,0.09478117785240946,0.001425263997741579,0.4138694055669597,0.2403744869197001,0.3939346099722779,0.468382283115482,0.004930912601012859,0.2518564354596122,0.0460458325862693,0.001752826771191309,0.2259140439902251,0.497723891683111,0.1804691871521636,0.02980222963852628,0.00693899492379056,0.02400332108488189,0.1902910640637322,0.2193316253953126,1.353544586956394e-12,0.1796664403982783,0.0008236196582957116,0.6622583734953374,0.2760016192838565,0.01391533686936873,3.486710454101017e-15,0.008368221477989372,0.07721440671729286,0.01799308570539811,0.0103744052131774,0.005742901097721439,0.01107151995776075,0.6183520123477022,0.03488626534638974,0.5109526954674987,0.6760209877502729,0.001047460601154643,0.5441267241231195,0.1961454993750298,0.1157227334287068,0.0006897520762896171,0.8726294499724538,0.2752116638653727,9.869899533255479e-05,0.4323687212086786,1.325379141783546e-06,0.980730398142588,0.9658307727502787,0.1837076830554301,0.005616397470682703,0.001458935335479238,0.01768648864392848,0.06758827122756504,0.0004940356413286255,0.03158607888972702,0.7112758362984893,9.680187188119487e-05,0.006105967902275105,0.7764362240824428,0.01427204650314575,0.0002698210779690124,0.3581112192629415,0.03703043885126332,0.3035338770311428,0.02617838002740309,0.004857178494954768,0.1360006062054928,0.1736675656148155,2.855366409424657e-07,0.4468060751460923,0.0001589821391830171,0.8433509010467662,0.1044014605298139,0.8499843360916679,0.005767471694321016,0.7208103760153015,0.5192235235918178,0.02512549464124628,0.008579488391880337,0.8048275893137194,0.4681488411889431,0.08589418016077566,0.3930265803169479,0.0885108730206496,0.5203594528386332,0.0411011119700403,0.08980336404046368,0.3795913577833035,0.02640451912765498,0.4845758193367441,0.07013747620195576,0.8545619838096901,5.495476490248281e-06,0.0002676434748535906,9.295787766793223e-06,0.7901995803832508,0.01651810441898322,0.004110101485071283,0.8753043882761575,0.1005452969173776,0.1825112615828148,0.00627772416583666,0.07929702170106022,0.6695902308065332,0.06503825543733412,0.02246324345771112,0.7162986200309922,0.02131434025244461,0.1769440922327415,0.005085826763973279,0.09744901165266286,0.6248519765367727,0.1646913381789313,0.01412877003622879,0.3746289456233961,0.6405766724402243,0.6056713866539114,6.422290292951179e-14,0.531362016349221,0.01229369159040735,0.2344710946410107,0.194595363050285,0.0003161436621708044,0.2231134982955826,1.01737402720683e-05,0.7263926520757243,0.1181245346782153,0.9175763090243902,0.3531521943779407,0.9274879949029972,0.01343488534289271,0.008614060291610416,0.00163895453560344,0.1704134269067686,0.2873025166759224,0.005638814247062336,0.0004957954356484764,0.2410619172278046,0.2301241469857813,0.00662421606936038,0.5255331836314646,4.487307167745775e-05,0.1566237553056567,0.05019938689395505,0.0007521768122751422,8.002489687642771e-08,2.019623153701495e-08,0.02686381057576108,0.0605006001513658,0.9551468994914727,0.3617068993264604,0.9280198536998974,0.02957526006655822,0.005587634364030853,0.01274658188213618,0.006636899256255911,0.4362759861078904,0.3081560869492858,0.0002157711310518337,0.9343034518522337,6.355486036557814e-09,0.0003822606711597746,0.0006185185164508632,0.6175124774463727,0.1104478831199934,0.562087151306611,0.03581359765679977,0.8973324923681029,0.4880311325945096,0.002995283459678851,0.09566622708569009,0.8227056143931116,0.965412557687329,0.9793941529043421,0.8133436990335698,0.1009646148178902,0.9838479759916177,0.01250082135308388,0.02871879288197602,0.8032017338007011,0.411171228376626,0.639120600963225,0.3660502863446217,0.5396109859119631,0.02468686274143579,0.009722916813833736,0.1852551810815975,0.6620711622399017,0.7144291317971496,0.4553486387885791,0.03754363097437503,0.0001051449211563538,0.7606470020408361,0.2795761266315284,0.2100830454118319,0.5211547617270254,0.001983227096016814,0.2610291851583873,0.424880409371561,0.9384217152214763,0.001504512106308413,0.5338363400441014,0.0004017178350705403,0.1571356974680619,3.102593367896112e-06,0.4245644849051275,0.3347908322928776,0.1656321263612232,0.297879868460746,0.8448825728047383,0.6455458823485475,0.0002072722368713514,0.7276153334763976,0.700967343993195,0.1587996901428861,0.5926163406601845,0.7080888933366347,0.2711580328389318,0.0002072722368713514,2.783278994374385e-12,0.8460126507081173,0.3939080869435996,0.5561074902788832,0.4688547355342261,0.734738656130304,0.6734434660698077,0.01493221978209885,0.8025772152504561,0.0001142270750379803,0.2643471155973277,0.7841610591431083,0.1184203606749371,0.4423186812962878,6.642908943008256e-06,0.3097381579779688,0.3702844823523813,0.0001784883869374758,0.5896926863515476,3.707298890351264e-05,0.0004122727943895652,0.002484355701312278,0.0003718667918918406,0.0894044490017163,0.4411953682597536,0.1661384182128173,0.4310248776162534,2.090320664569363e-10,0.7640007914971797,0.03909883248513071,0.7465061944804386,0.005314062168272451,0.9474643564167003,0.1302931641814568,0.02336475945739358,0.6323184636918928,0.7366778609783335,0.8372420849211373,0.8292947823189778,0.8071879435803547,2.177477058444724e-05,0.002910043713561253,0.2163371822853391,1.242784597672283e-14,0.6926157488206697,0.1935204398333558,0.9112880290507469,0.6590773652780622,0.0132759181726229,0.006710798574567915,0.003897224041593716,0.8951236248189153,0.3879514270520726,0.007772235881474382,0.01029771902108256,0.7632865760377388,0.03201890900449889,0.3945290596467932,0.1210965585570777,0.9701384356845415,0.9993679766352331,0.0002058725263518021,0.2709491026600299,0.01430821804026352,0.1736273163333026,0.2279731521927236,0.1062169242110432,0.3196123185561531,0.6206718567078529,0.5027899826709997,0.01327410983310375,0.01507029168619027,0.9028178605484292,0.110422123293955,0.2237282101570711,0.2919089362576852,0.07955890371153386,0.1046002953650664,0.5379975116639537,0.685477989836521,0.974586141396193,0.6404066323888823,2.233714219142448e-06,0.5999191167929804,0.1249045378291923,0.5449014864343116,0.4344033913134037,0.7969189055004233,0.005646137123286296,0.04592975622056553,0.6411869779440694,2.364649774427726e-12,0.05557291285552907,0.4318984007727337,0.8007150452007552,0.8138085570195965,0.5004490191695895,0.8415730103256502,0.3837466323899458,0.9463285431518763,0.03142778507934131,0.245286585733528,0.5233862624381322,0.2296466806671116,0.02768107338885653,0.0002861385006073048,0.08642988491746671,0.007513573946785827,0.005919942402112065,0.0020135154816408,0.6216195104712925,0.7378739690915429,0.8448825728047383,0.2117662371280497,0.01524679619072917,0.8182751965414761,0.0001100220510859584,0.01464699068572493,0.5184576264854017,0.3480438662003515,0.003439451433775311,0.8451735718849173,0.003680093764058697,0.5561074902788832,0.7071216716699456,0.4040321162379268,0.01313968670295229,0.2778801261509926,0.5396109859119631,0.2767368635144892,0.2841579195323632,0.3694618573834348,0.0002584856312299216,0.8249671407175572,0.001983823577418618,0.570057059373613,0.02267192600307022,0.3003285287270344,0.3318602978351874,0.9257090085193957,0.4590763089654884,0.1191264320718183,0.9622502650958906,0.559216526261659,5.986966522095721e-18,0.3672475262888054,0.05202115966333867,0.01910572269556964,0.002679656319003659,0.108209638252463,0.6063311083079138,0.01240880994299362,0.301422981300879,0.6045017014769808,0.04341802301559013,0.8714900036831484,0.0001100220510859584,0.04451858365277369,0.1059478688878273,0.3954030111422409,0.6576408755314127,0.1833868396537083,0.003152546079913842,0.5999895952100606,0.5417844250867309,0.2797290625839597,0.5551414806356442,1.654201985414966e-06,3.951133892568758e-05,5.762045013279111e-15,0.2563096007878504,0.9355986906707847,0.5630513185045434,0.01365075899240884,0.2071418473614403,0.07422839808718243,0.8286355668977962,0.006132063840273053,0.6944356402962001,0.01638300089390101,0.01583746591414059,0.6943361027176257,0.4245644849051275,0.004581261536926697,0.06940161945020498,0.06322568806020396,0.850888353844906,0.5542963600044858,0.1785711633599749,0.07073527597331367,0.3008344825475054,0.6933893212921673,0.6141965272956401,3.939259330507901e-06,0.001950859212750752,0.09014125857014889,4.922308419633358e-09,0.04753259149322114,0.1680734831816569,0.0608268173010035,0.3108617691823833,0.9299394291193157,0.02658679619466104,0.06888623350667104,0.02389236755736205,0.6392475692659517,0.01964553979376892,9.890408699859639e-06,0.002130673274901875,0.00665828734010428,9.115526614404055e-06,3.347044073554325e-06,0.08289959046194885,0.406951017706053,2.859188829060773e-06,0.0005362436003414164,0.08227132477383015,0.04887075103256034,0.0233478242513074,0.6959281863595775,0.3372096548425262,0.8811348616346973,0.01430706538712215,0.8398852233854034,0.02147528067858338,0.4403399767047642,5.623290842995151e-05,0.2142649936740288,0.8080565694956535,0.001447096555084328,0.0144506273449295,0.5085737901228793,2.815143559205831e-14,0.01894207216103984,0.4593410357649612,4.544442209102549e-08,0.1094012654434308,8.73881580336644e-05,0.2637527487155875,0.0006625190077894625,0.6777169958663473,0.3504544547096465,0.792537497716813,5.239745065040843e-14,7.796214267270061e-05,0.001605795218436625,0.0009112455380779819,0.2044865846997752,0.4507015566067142,0.09068791643290566,0.01309639465968686,0.1075943439902602,0.01129838175786204,0.001996349598030377,0.000386669302578497,0.5864327113825107,4.576228830497498e-05,6.466979190379348e-12,0.0009112455380779819,1.995276080859568e-19,0.7829692509764705,2.918495366836507e-07,0.6649292228424223,0.6024856702234119,0.6088004503264968,0.5279903579026678,0.4789639021451162,0.0001725153452452688,0.9108824286521974,0.001370823533371143,0.009447216629017352,0.9139044406535622,0.7662222849825863,0.2448024753040467,0.021589387992025,0.009335007772597778,0.3871986268576647,0.3788625730273884,0.4411307403649212,0.1726833506479545,0.6593971905764445,0.270898440391603,0.4713508812069368,0.891131492131317,0.3698086207145589,1.699869846766803e-05,0.1213973556689378,0.1626865584745813,0.3339413864390708,0.1115261141074456,0.2035512209712131,0.8844118236867099,0.8951601911895556,0.1475409608275998,0.2824533027873884,0.4646021802001435,0.4974510287024171,0.04667220099795701,0.0002861385006073048,0.000138294215091426,0.1891530990547475,0.9542206217321727,0.5887691992025091,0.7739831541730282,0.891893336614878,0.7399816062853595,0.006738316371206934,3.385452695690157e-16,0.0354199211597784,0.0004595660373716189,0.05682823175391329,0.0001771567266043868,0.06708820187857387,0.6614459764256787,0.1559057244637244,0.3131599633749981,0.4558818558778414,0.01890522906282292,0.05846729403306523,0.8690405171724176,0.2115111233166761,0.05531857069895739,0.004593583649167145,5.302418957157623e-06,0.0004627167964433936,3.655934289029617e-05,0.1239705729170829,0.5735697287925307,2.205396870948807e-05,0.004466904243157043,0.2303258829514211,0.4694909131082061,0.6498399416086346,0.3663647774403387,0.3753909398265928,0.4184237357692526,0.02822334527571599,0.2419117055200958,0.02592271036449623,0.0002698863273626864,0.00928509540899502,0.4696886641290403,0.6279536236026422,0.1303754076590176,0.3653892134654438,0.9022914913695191,0.3084614770411561,0.8916324665970814,0.4337904927164962,0.3582739852061717,0.04835881830385124,0.06084516536842918,0.6008072105131704,0.06046483284210526,0.000633707481717641,0.09752566935571129,0.3876929978116246,0.009383780269105449,0.03837932682729837,0.0122414148157172,0.1829938639314384,0.2817799357280833,0.003255653928441556,0.01522900510267673,0.1567864638683652,0.08670043917908178,0.2402952953718569,0.7068768104649273,0.1681108977739521,0.3738456356191995,0.3628271058108801,0.04080846132829809,0.7171091001765169,0.3203980842720065,0.1175290593059779,0.4199136390171966,0.04351214806248967,0.8767544402303131,0.5681377515759917,0.838281589565302,0.2303744305866311,0.08042515974539252,0.03541572974991097,0.4576950703038906,0.7408635786055862,0.226710797947595,0.3465820284091374,0.8701980139756318,0.480134553061943,0.0002012465528880593,0.001819507967065722,1.082180723278036e-08,0.8978938273700076,0.09695046356398258,0.0007112882394745045,0.6499039116352138,1.494131519025239e-22,0.0007081999017261635,0.007993530656745441,0.09891844672954468,0.3890853207609919,1.223828557562679e-07,0.2312749923693553,0.5210963633468754,0.8932237122568596,0.06674332932227239,0.3581969906366797,0.0260231196677523,0.1696794863315639,0.5246023052305091,0.8636916187733974,0.7012058312104185,0.6967971108665729,0.188807112394463,0.3756515706443023,0.6635333271430225,0.003727702950711503,0.3620444480145534,0.6498399416086346,0.01602047162994467,0.007823434608484222,0.0001889433313776612,0.2325441761910928,0.4470649994786088,0.04738732218765003,1.066799460299128e-06,0.4730050018319579,0.4881096438821231,0.009320385307665888,0.7878483052400369,0.07222341160940636,0.8760636646201442,0.008316460197520413,0.1029928459595991,0.70558544842823,0.01579326849474933,0.002634042526428928,1.521878605082144e-07,2.448302363229338e-10,0.4078352652560524,0.0443653841112262,0.8930210967687493,0.4161207167586372,0.01197203632942716,0.003805357134250072,0.7893654672170818,5.596250553157659e-06,0.03382068910615031,0.5099027256609273,0.003723430450228658,0.0001348703526939568,0.4783821806507634,0.05998252334580743,0.09966235153942408,1.070236310550331e-05,0.05263213473818302,0.1250914050744967,0.0003751366341335338,0.04191808752162823,0.08016843880616709,0.0004994807747069784,0.4999992635939156,0.3876122587609715,0.1088655460931745,0.0007180226416800857,0.8771383299840289,0.2868002882890875,0.001729071339403885,0.00393118363730046,0.1602970845829451,0.0002519333426681784,0.001520441536513738,0.4910362831962745,0.5037330436573185,0.04235830823967331,0.569645283850519,0.03202366262028445,0.5142771913772898,0.2875491386838063,0.2082560051731769,0.3639993399288131,0.04671882941337673,0.8856542052186801,0.8874594722922451,0.2896541423252139,0.8833948363729157,0.0005028512892319098,0.9541085010376312,0.6869534593644449,0.07656807973168119,0.006414840599689012,0.03940458842431839,0.3655393200830152,0.008988709817766297,0.2197108703024903,2.434578856502639e-07,0.7813552650888332,0.8237502198977877,0.3420391618870676,0.009325869519132226,0.001240799391364046,0.001824624374381683,0.009611439721511161,0.0001225212571405915,7.250436574847088e-05,0.09254635138857564,0.9944945315157215,0.8945839571098519,0.3113002272331163,0.8895988707032888,0.9803073240467792,0.03030512841793643,0.8653285844277752,0.06888623350667104,1.2656129351496e-05,1.643591480563512e-05,0.4271229537642844,0.6270576945222986,0.01760237641815095,0.3277496035721955,0.140765753760884,0.1360629914994124,0.5619217534158607,0.4988337298157328,0.09330606292358848,0.8004194475165434,0.006995718728058282,0.07927123893172458,0.03198294604014064,0.01662948891752048,0.2149855025756453,0.7324749388062729,0.5551414806356442,0.0395468775424407,0.5427033963107311,0.3627225537163156,0.4836684503654015,1.007826046300852e-36,0.005950595782048048,4.621448912534173e-31,0.9340646862996124,8.003259779476067e-05,0.001574166741473978,0.2301741801563276,0.9534553094741761,0.5779383917440777,0.07768689748366496,0.04591395021018074,0.9658307727502787,0.1236372305346916,0.1063436158854833,0.04808762744273664,0.01636025982397868,0.008817480416236156,0.7060065879418361,0.02779160271205177,7.113372744842397e-17,0.002098992236046552,0.5379975116639537,8.324145351458829e-09,0.4420509286511208,0.7909038092322691,2.307861316290088e-05,3.508827374419642e-06,0.1436681102874989,0.0144506273449295,0.01115949087821304,6.3088899991362e-07,0.005136428819516458,0.001234333363916094,2.744197682454452e-09,0.838281589565302,0.01949734789065248,0.2884894789316464,0.5414620756962666,0.4776883256058933,0.4735987596572998,0.2098079965167439,0.007531816919219041,0.1466790974168839,0.4506746095653185,0.000163594241459608,0.1111165989005177,0.8122777077752956,0.1601426093312325,0.789208918435985,0.3788735004403594,0.006680285308890083,0.124726721831177,0.01250082135308388,0.0452915987926069,0.028238820575277,0.0599855000045691,0.02679923785076443,0.8636395147993956,0.003311761025680209,0.07162142654879741,0.1041483374875236,5.194281333831508e-08,2.836202779970986e-05,0.2050159490081888,9.586192055710756e-08,0.02445382970670921,0.3809648921274866,0.9446197617861039,0.6354609608466096,0.002314785835232334,0.2996744623872043,0.8811348616346973,0.48533191951715,0.9590397643373596,0.1954679939277215,0.4467209718544032,0.01751760182320606,0.8497249732880033,8.933384926180463e-06,0.04909196993762387,0.00772355284413007,0.03046208805003027,0.04476851553240652,0.2051724846331411,0.1336684248534345,0.98922824188783,5.157613246554566e-05,0.4411953682597536,0.4213268252103292,1.300172301061273e-05,0.7005263955199341,0.2140963397224538,0.03195704109872479,0.008305508232476316,0.8843249868668023,0.9126875890545249,0.336113067407865,0.6872663788178244,0.7367169563802767,0.0008680442411822454,0.5248999841230851,0.02389236755736205,5.182084643115293e-15,0.8200546315608412,0.3148849365951662,0.4040906761865394,2.910742947315268e-06,0.3945380591383901,0.2402685215641828,0.0009995841042082054,5.981338639802618e-18,0.02065134302710036,0.0006777744954609748,0.9933703524580143,7.319790239847599e-11,0.8079639234675328,0.1808310898366843,1.754600328655514e-22,0.1120090556163987,0.001719343789911347,2.542159901067146e-07,0.8460126507081173,0.782827271638073,5.974892787488696e-12,0.781181733888523,0.002563144464739248,4.931430300776283e-06,0.06047850115145127,0.6360678154152595,0.7467078074997224,0.3844207978091382,0.3264513171104193,0.01517512296631817,0.6939780744987122,0.2001021492179154,0.5962050333913168,0.4387806349793882,0.01226527247538659,0.6740747776442819,0.0002113544187052447,0.2065924835063631,0.0008918545734108556,0.4290037898529719,4.680169106989102e-06,0.01732364202919251,0.2390777286238971,3.828881859499525e-06,0.004996775595930654,0.9457968926325999,0.000522854339980184,0.3187559081783324,0.7913946718773649,0.0005421554815845596,2.177477058444724e-05,0.02094416593664142,0.3820512839635948,0.03430869766990163,0.5438293970391275,0.007157009424752183,0.0002436913432139599,0.06013902142871834,0.9051486614216956,0.000417572353428751,0.01357891605461999,0.333687110978711,0.01142763190987285,0.8799442430489973,0.4098614998144098,0.5448284721666843,0.04025926498315386,0.8467527474349713,0.4281175743074068,0.06545247478393336,0.1620981003624161,0.0004652423750396078,0.4318810840625703,0.9677657230095952,0.7112758362984893,0.7528266855525225,0.6305638530153973,0.4882107802911305,0.0226795552908435,0.6871359032325965,0.003755725567125795,0.6022227502843615,1.352007994082651e-05,0.002275341228762049,0.001148808467676792,0.003112167503474466,0.04299196880411629,7.54035708817951e-07,5.205814154017283e-10,6.772695492299169e-06,0.000480985911688747,2.39832243048258e-10,2.420287774780931e-08,0.7166630783696201,3.367145651703629e-05,0.3556887164671298,0.002511847705208071,0.1020645326573121,0.242253856594513,0.29898976558949,0.89530380068149,0.5715449933825525,0.009577519728309239,0.1340829687365651,0.3004949152242352,0.1512725400824372,0.06239261787090515,0.632466826822056,0.4215040253342953,3.28722963436648e-05,0.005079043427902147,0.0002209436920092479,0.1156463936833632,0.7594987774691228,0.1686886309487617,0.0335544465672803,0.253925575178888,0.4411953682597536,0.01671433775864658,0.9527117666466172,0.9177166280880124,7.434148658456207e-05,0.01546269955822814,0.02082790707227924,0.2963702150339146,0.9731341689955082,0.00156463389190726,0.7772542237249117,0.008078408687084905,0.008851625154453207,0.3735522613037222,0.49449673965009,0.03737188103818982,0.1602621605539953,0.03316897802820055,0.3834822815232473,0.09893739458381987,8.295584373449469e-11,0.2169817655726306,0.236165164392358,0.007234802113255978,0.8298303192282441,0.7323860466692993,0.6882945004850091,0.4932016380323925,0.4102345146421611,0.787441605418491,0.01103737127275245,0.9390323771213417,0.3245345406011998,0.300951553352892,0.2841515104345309,0.000953028585972773,0.9216119001427022,0.01261190652873174,0.06475509165579951,1.735117476480303e-07,0.01982204467047962,0.3761062153511685,0.2395648003504091,0.8876333576935422,0.001041329995183889,0.2617038975645593,0.4918781562733703,0.1467112217894593,0.65806568325855,6.043409265307203e-05,0.001445212720127164,0.8003518986397525,0.07066050323893329,0.07496613678501267,0.7096108717234677,0.09429830506687324,0.196377786432497,0.2173368226349943,0.02038141644153294,0.0005113337207999193,0.007281898630522057,0.319311701515297,0.1335765361693688,0.5569780421042256,0.05922837191573006,0.07025620239207593,0.1551490078248739,0.4093280303962137,0.8467527474349713,0.7738352152207233,0.4640012449707358,0.01982061184537801,0.08503004056970764,0.0004162575756701545,0.9462605856800654,1.75410065231381e-13,0.9511877420567988,0.1044014605298139,0.1365927708655024,0.5930744002589982,0.1021657915217773,0.0929310711492032,0.0002002437857167031,0.8226723760557845,0.02723360620751791,0.00757586057182298,0.02167623425386292,0.2779861404118119,3.603216806535564e-16,0.6447659665917317,0.02225264588105353,0.06463215933264727,0.01645960961160011,0.965386520611962,0.3502025937301083,0.09996179528285454,0.0424053789023124,0.0004425570286934647,0.6588857542065193,0.007229043296294725,0.006310060478989659,0.5644622471201154,0.4568137471072723,0.3715955131912994,0.3475223102317266,0.9143856868525048,0.2207607351600009,0.04191427028279173,1.290258648316378e-11,0.002195832834098739,0.0932403106397955,0.001180618085500503,0.05598242782700749,0.3309533116538398,0.2574950510595604,0.1097575162918341,0.4076908855545247,0.8909365525710222,0.5112312557559812,0.0004586139596230963,0.009569206768556928,0.1310715824786871,0.8913118275842538,0.01025412018773673,0.2888399186032835,0.0001241163776907808,0.01021003227964612,0.04741265796328924,0.04742319071166129,0.9143856868525048,0.008276605375244829,0.3111957910769586,0.2209920501684925,8.28237070557296e-10,0.9316470433649914,6.556826590634355e-05,0.05645950021707712,0.5775936182589401,0.1023051164225779,0.04716386875929858,0.8767251799444022,0.1623665805573374,0.1529649524825856,0.9260135068371551,0.0007180576860729308,0.8518865819123895,0.07314914857618378,0.08664866675242676,0.08116267210008749,0.4836393698051709,0.001568741138783485,6.475506746257704e-06,0.3580896165791256,0.3759647572782574,5.100915257684829e-09,0.06273507566830125,0.05869898837617995,0.1197680194843393,0.303674269675426,0.2869260348802877,0.4864366409855672,0.06593399206737653,0.8461415551112957,0.980324187467954,0.5484453187756929,1.363389636695517e-05,0.06468097576645913,0.151364556445672,0.03425921594952096,0.2235556730689,0.891562178225777,0.03257472951862014,0.9275369685350772,0.6367591541986666,0.3197675688978915,0.0007482413945119899,0.9119572903920141,1.076910140511117e-10,0.5309267315682344,0.2647671271064567,0.05970649615235323,0.0001457689790437158,0.0004217014877127329,0.2574866015437735,0.958434954909163,0.9553308424446226,6.542031605602878e-05,0.4824068951707363,0.04161843996483466,0.356124083662881,0.8322455007094283,1.802995522458583e-06,0.01957044578654195,0.006720628512003459,0.3772892100610103,0.4395477240900566,0.109054095252291,0.200573894326711,0.6226361927857866,0.2662688511373112,0.5035777996887982,0.8317951071684306,0.8265026479429289,0.001080287918352472,0.002579907786712969,0.2301546213402208,8.469190791960077e-06,0.09848218426066327,0.1096358901062499,0.3162906002200685,0.05829713078306428,0.2852210146417447,0.1297818138614708,0.002664675851450305,0.000106312879699264,0.0007092575811783457,0.7034198687354468,0.1166679000335119,0.006842160771842721,0.8870890743442963,0.9563938413518758,0.9703598155592029,3.862422394832335e-10,0.9747822548021569,0.0003716348929678494,0.1648424912747919,0.002903780997150368,1.445052264555845e-05,0.8084403921553645,0.242253856594513,0.6618062419101925,0.3285478245559572,0.591092588756965,0.06902080450320901,0.1503743007717031,0.004541751811952942,0.03335139335948647,0.1322434125518213,0.08907739503722087,0.000522854339980184,0.001722169493631149,0.005168708017587025,0.002172393939428125,0.06322568806020396,0.2821946287292284,6.108565508200775e-05,0.9463285431518763,0.9528871196109612,0.1762908394853089,0.002892637858106992,0.892216996241968,0.01100590163823148,0.2035870258699891,0.001312259153322,0.0009161541413066083,0.1272382985321968,1.896511003868862e-08,0.168337489243471,2.718515037469353e-09,0.2610291851583873,0.9855474827226861,1.207408318836557e-07,0.9203245028968917,0.8898768534148275,0.526308112805806,0.005084425267645528,2.345523390989491e-06,0.005088425893915769,3.832413561558955e-05,0.001702164927675827,0.0002786347208648927,0.0002973911828955566,0.0004175166667746642,0.2310078121501978,0.0802058040995698,0.01735686767191923,8.697341755122459e-05,0.0005406958221652452,0.1232603844781485,0.07816963487953042,0.4016057074914142,0.5232753206357204,0.0006439906097742759,0.1606000607473106,0.09249757476310387,0.00203559908716307,0.6763610439148934,0.09201374553607097,5.314330482301129e-06,0.6578620898078757,0.193192181653711,0.4654348305047873,0.3275706569446336,0.003224386178406124,0.0005318410133138608,0.3261304314139188,0.0005743913599894082,0.2338842195015565,0.007684837015318814,0.8589355940303313,7.197189986218952e-08,1.759051116104892e-06,0.007478849930196485,0.4124000227342511,0.03677778952519908,0.3795311151071906,0.03215205727425906,0.9931593647709804,0.003188328914247866,0.70558544842823,0.6089403227866481,0.05702569723268602,0.01360633838464384,0.8260024114821645,0.3354645011368032,0.2395648003504091,0.00935043273330481,0.0001192148694192667,2.992162542201738e-21,0.5144566356160906,0.3866202269640096,0.9107085811692675,0.1028451157856982,0.5715449933825525,8.348218680946139e-07,0.01198104690033338,0.0001165243103226693,0.0003749370217541746,0.001596695018377189,2.960073549591658e-06,0.573950059264877,0.00928509540899502,0.6618988228559839,0.3598861891501237,0.5417844250867309,0.007493413680081554,0.1796664403982783,0.001344655774748403,0.7689714695996296,1.785938243156075e-08,0.0003016308172373518,0.003719528791995494,0.03093728858443858,0.9906851930146899,3.818727111631564e-08,0.8919277655371299,0.9395421267437172,0.1174024717794584,0.2529698832460578,0.0001641254275596805,0.007572087279074079,0.0001892044656934951,0.1073656792100171,0.8582869630560805,0.1282312800058442,0.4040321162379268,0.09597440790296674,0.253131380734831,5.449857507789534e-05,1.539710759784665e-07,0.3761062153511685,0.8487628549431723,0.4797213122813993,1.522082671231949e-06,0.3465820284091374,0.3276158663941455,0.09068791643290566,0.5989686596939934,0.8833948363729157,0.02409941574067156,0.2125500839175402,0.08957303269417602,0.6926157488206697,0.09176367081018723,0.2942686366346032,0.8422800845997107,0.297088849026535,0.8568862452067008,0.002910230550215051,0.04592434130548367,0.003017370380978555,0.0100865593070165,0.6526129853827436,1.199260614929593e-07,0.07985815634836668,4.766180711144957e-31,2.17522790308388e-05,0.3531905535932645,1.052176219946115e-27,0.0004187991541253386,0.001646934755805004,0.04677890469517033,0.3517851446938847,1.220964575529081e-05,0.6276419580159607,0.5192153568643473,3.13189299625042e-08,0.8451735718849173,1.419161425531828e-06,2.452102330604302e-06,0.01199289156651527,0.6802901383517097,5.225938785701028e-06,0.03577266395027816,0.6644636377438621,0.399517507517526,0.1080460764333221,0.01893186370999485,0.8370957352427816,0.563654875639284,0.05286563814801881,9.115526614404055e-06,0.3055595291447221,0.06818026995287874,0.3809250405798698,0.601736551744219,0.5848282711831043,0.8785619564017452,0.001552884163662162,0.9112880290507469,0.766016809179803,0.023589768842295,0.06360017307048013,0.005339690371195848,0.002579281061954985,0.3091910303559681,7.390289181795038e-05,0.02121145420026619,0.01778196322249231,1.377126045591202e-08,0.1567864638683652,0.5602782610242212,0.538656924403043,0.02147528067858338,0.007136998682003115,0.8897018647353522,0.006713668387184423,0.6468900313758095,0.006127713967580548,0.007482977171824762,0.001098676101647448,0.8318225256930024,0.18561994135805,0.6971943353981389,0.001539236712265653,0.6629571868009061,0.9901999461317573,0.91142215096657,0.1806414185917114,0.04124055277487652,0.2442690119514521,0.3414203850830169,7.227379032858128e-11,0.008288065635184097,0.2100830454118319,0.118761966452148,0.8469223328393746,0.7012804476502231,0.006704173184392406,0.05907731842215125,0.0001353150295067844,0.1650024038258021,0.00464441189643192,0.04996899811829311,0.003307821540294058,0.03823413748585337,0.7095494793164492,0.001386927598850254,0.6677737513523793,0.006370209167242997,0.2216276061323531,0.7754118105410441,0.5791055727635608,0.9335888155032672,0.9237858916549762,0.4320657611465989,1.552487643869277e-18,0.007443517270510934,0.03530901552221442,3.361269460333102e-06,0.2991677187595757,0.0007241026013371931,0.01044257134834823,0.1260744143843269,0.2533403156194551,0.0153085840894762,0.7339133576680296,0.0809038536586548,0.007905132436802912,0.0591735001520866,0.6278371808528178,0.0004967542815586822,0.006130123820283028,0.615359909322637,7.031187890823752e-18,1.56360374882643e-30,0.7751693448968439,5.314330482301129e-06,0.01717027530880686,0.9525597027122085,0.03129670624187745,2.865395380390998e-11,0.008361952376614381,0.3479837871319909,0.05048861853437588,0.6388247999253228,0.0002955435366051787,0.03546674624863449,0.002356156561595349,0.269045783837439,9.656847236693502e-06,0.5921095819182535,0.03098751756722547,0.0225067382351514,0.0354736409104148,0.9568011634313308,6.440042307580911e-15,0.06084516536842918,0.4526922182737937,0.5574149501770199,0.005501292294714791,0.353185587113322,0.508071679945365,0.01046928365039541,0.1507418737821729,0.0316512876645105,0.0018874150177043,0.008093929101220889,1.90514471840878e-06,0.01616255379840481,0.2919140723552485,0.02611137473702535,0.8513414482102312,0.07505439098056518,1.266250363132785e-05,0.07709938492717801,0.2911573476949791,0.01971132750007707,0.8541729810136108,0.01971132750007707,8.938713790431126e-11,0.2750805263766428,0.3411727283693062,0.7943193063323649,0.2206826535368717,0.03856392087035785,0.2916306991176797,0.02768107338885653,0.6253921959554808,0.7194582513178887,0.0360856928038147,0.5112312557559812,1.488359050418319e-05,0.4874189730561954,0.792537497716813,0.757339459998293,0.0281794462023632,0.9368770779396367,5.417933232485788e-05,0.006035404739274068,0.1989889081145306,0.1029581711265275,0.00926048272986432,0.01133518530086121,0.9567280045814528,0.6866755084317032,0.3681987597739828,0.8277799026789704,0.8843249868668023,0.3499255536854569,0.05641461290434854,0.005265217655995512,0.7209613042191755,0.04143815045163816,0.07748104894763669,0.2457097930447608,0.8513414482102312,0.8932661959195628,0.004764480588820816,0.8435591650826673,0.1453484793774483,0.1209963791922925,0.459446053507102,0.000265503562944247,0.0216391985401488,0.01376833595767797,4.682517665498075e-08,0.9452184491573717,0.01734833027838974,0.7171091001765169,0.487460974783932,0.1274310119796887,0.645792138833635,0.0006096791927551522,0.2906833529567041,0.01968087676225777,0.1743876155692449,0.002130346997068722,0.03610022315070915,0.0007907314423514012,0.5568170544229781,0.04376923653339924,0.04631864887431633,0.0421708321575214,0.597367586257916,0.654734577653192,0.5531274538491123,0.002405020885403631,0.8292947823189778,0.09026962532216276,0.05868335982380034,5.786341490003397e-06,0.4338999576573053,0.368349062672701,0.00126488405785204,0.4363352453181064,0.0002698210779690124,0.9444635942052373,0.09274559460724627,0.08106956786908597,0.0007412122749015542,0.3571762082415687,0.710967294220084,0.5464759872194823,0.0001227738248889308,0.2027019604493007,0.237407939969096,0.5019133800651832,0.8259940974913863,0.4932016380323925,0.002166158627729522,0.004231103656046432,0.07394730252376103,0.002553703313231723,0.02315918978481053,0.03423998864184444,0.1827060663123524,0.002473683484392214,0.04089506320823597,0.0001658985226807398,0.8138522230229602,0.5618063946485908,0.6504456818213786,0.02508388611835065,0.2152729740900717,0.1426295169187782,0.3653093406967555,0.1107646214155589,0.685339599623771,0.001685372018691095,0.07434969316719428,0.9964906732910624,1.240596332820383e-05,0.4172588617886501,0.1714802331971429,0.000311030209478929,0.9138217793314007,0.5505346200146731,0.2650826923937533,0.4139829796888281,0.4433267086368337,0.8652216524665488,0.3414476764018213,0.2933884825668035,0.1891530990547475,0.001069918571306415,0.6996755030148148,0.04599506529768682,0.3789512860805644,0.6797073027955288,0.8301357898732277,0.04481100659945722,0.09893739458381987,0.8652400748011406,0.9087222719810929,0.01284312997894173,1.070806040376512e-06,0.07485398860668793,4.899463450389374e-05,0.06258632530041852,6.73634735882661e-06,0.004437200312728069,0.05381719188289224,0.001022064378687996,0.2486446748876618,0.001997664450270791,1.731976574528813e-25,2.396424601596003e-10,0.2626737613994291,0.7894776474325458,0.4947075002767427,0.05846729403306523,0.2724133980359639,0.4851509326618199,0.7456781815562178,0.5998001704913124,0.6039436971717954,2.949408240893464e-09,7.813453818949301e-05,0.0001698750859196168,0.0004577385865399685,0.001734152722706868,0.6199599932367441,0.02497109169653103,0.4123379756873115,0.1375135556551126,0.3233896534521074,0.7601932471678808,0.7998490150285681,0.08029029137440856,0.7305521761820414,0.78043312662868,0.523298179852063,0.07229507249510861,0.007793477753210469,0.3296993141850661,0.05924648455655371,0.02720969166800767,0.7700991483943982,0.002443733181812692,0.06203119334992729,0.1332781625496044,0.0002533380621561138,0.2486909305190843,0.0001181996629717561,3.876557398597472e-05,0.002843808079380429,0.003539989000976861,0.4975341231006372,0.3190321753072498,0.8174652706922432,3.346000245392561e-06,0.0001205571743233071,1.285088568157821e-21,0.5163420177728958,0.003828318132032211,0.6802479151027938,0.04223781989187476,1.350657945088956e-07,0.1261615048491791,0.6370097012256013,0.05934984306546071,0.07763486368085555,1.760743818409128e-15,0.07371285613828059,0.1787571612398791,0.8322282517750902,0.3566219059235985,0.01115949087821304,6.434906773659445e-05,0.849808029522032,0.008354911578294207,0.00111733900605369,0.4245644849051275,0.7360360591809699,0.6137170234203078,0.1374109126563835,0.07642642584714918,0.7937707932839152,0.8434279721832743,0.5076244743430983,0.0590204762506843,0.8160631486596012,0.01645702948065339,0.4233682115645848,0.1434092667930201,0.4271612502601277,0.003825178995027169,0.3437373088567399,0.4576950703038906,0.5742519903763846,0.004835597102945544,0.4716189406116641,0.0005233352135941335,3.21181969325543e-05,0.008993692236169996,0.5753415958867029,0.7224605001415846,0.7309414588124125,0.4747970770255354,0.2780556571160933,0.8955006654104856,0.0001180729614051671,0.5332945480625294,0.04617428495166834,8.295584373449469e-11,0.8838897882451781,0.9146699966056008,0.0004586987803778874,0.03533415713810112,0.00112025540997862,0.7095494793164492,0.2229751822553996,0.1152989311324808,0.01462046883464627,6.199264473071528e-06,0.6555437341014119,0.9024209723998637,0.4532270467971047,0.9670718911475692,0.9903810939588663,1.132887844675767e-07,0.9231471678743822,0.6852223659394953,0.736553369916881,0.3801234899277844,0.3165479098749172,0.7160603891490861,0.7570514425346156,0.3581969906366797,0.7085163116587334,0.02931521827484901,0.003060266478351304,0.8156517852362861,0.292867405192586,0.4433267086368337,0.2500421443148282,0.1692749099649995,0.9231344806741268,0.4242863564631523,0.06092051963419863,0.002216047859575296,0.004533083941245609,0.3667337262441406,0.0007929957716560948,0.6813679277809334,0.1175360664271995,0.9651116188333161,0.8843249868668023,0.006583774928261956,0.006587523505603186,0.891131492131317,0.04877614234018769,0.9154511691709356,0.01483920422578255,0.0005239305760724513,0.1194077324275913,0.4202704882091963,0.8450236998112154,0.4249880408643882,0.06519223199865599,0.4681488411889431,0.1122138113845387,0.3167243990651296,0.03142778507934131,0.8534011668798587,0.004276771457845783,0.300834137899591,0.01871341401564477,0.0003388528657218563,3.483557236678077e-08,9.549116741231101e-05,0.3891683542189918,0.3626720147261378,0.07591823910910336,0.8804948997870423,0.03648658380629071,0.6812832972102858,0.746679098995424,0.9425356132940845,0.7548384842579411,0.06872334694070019,0.07383806461828518,0.1351194078384645,0.5821772677164588,0.03728820110246803,0.7792930557260649,0.3045894415535041,0.006628473381915201,0.3115757121756524,0.1702413832885345,3.638648073223419e-06,0.1025595015719731,0.1290943333041645,0.0002672020061338928,0.00094432825205704,0.005185618185112945,0.2771286346122904,0.6222062788320363,0.02840409051556668,6.570632799135745e-08,0.06465404063835099,0.7757671610805296,0.399517507517526,0.9034137997304608,0.1451666122240391,0.3188512764379735,0.5496328299364652,0.1903936901366286,0.07721440671729286,0.7887565809043053,5.538031176197655e-05,0.1541334162323386,0.004169449259501967,0.1178947682329152,4.493492429876017e-06,0.01335188092563164,0.475335879883388,0.2804337502015505,0.0003483764961564066,0.7378739690915429,0.02569872303724562,0.8188129905540558,5.991661360705072e-14,0.004161714510503125,0.005771089579970288,0.01450252798262089,0.7084423013421286,0.0002511785989549661,0.2626737613994291,0.03235987916393876,0.9524919743482566,0.9493242900952302,0.9285995070563278,0.9638770116791163,0.1819280533319041,0.1013718710992879,0.5043146341481473,0.01177451991069615,0.2495619754015055,0.4149560420439384,0.3625016212859888,0.06180144681502703,0.9735379407447283,0.2565177072534731,0.06577259940531893,0.187368749174264,0.7365253502620531,0.004688409143540985,0.0002175876661419393,0.9600185249387411,0.7786458609096681,0.3170218618618519,0.2512615833139125,0.0460458325862693,5.099326299296898e-06,0.0101825184595836,5.489325785083492e-16,0.04650879637959027,0.0001342826298756382,0.0001422970108490693,0.9412520158291096,4.915782321909259e-05,0.7236413834923136,0.3866202269640096,0.007888578028855031,0.9529412601810405,0.01413990053041406,0.001473610331439331,0.8312673128153206,0.0651119066531924,0.5092315255628653,0.01817083486196575,4.435567715933691e-06,0.4803278632788074,0.006043031162762616,0.2014339163123134,0.0264970734553335,0.1725796953569214,0.6067736062816986,0.4596148143771715,0.9121697085411467,0.4726769481391523,0.1098699090065913,0.1621235094557264,0.5517539721745101,0.5087640325179282,0.8415730103256502,0.0896953974437726,0.6855769847087571,0.08659031912047027,0.809105579617983,0.9623772842329845,0.00286623344827275,0.001676656947553678,0.02187469676990053,0.04012766105962029,0.02123119318934296,0.06272597719491771,0.3636293821699944,0.655828862665005,1.758739286009983e-05,0.2701958398682172,0.2041713892987258,0.1291876176155664,0.02382200740215572,0.282268904134888,0.6239337698957252,0.9934425175554431,0.0004445630716208467,0.1643212413830052,0.2627787850255915,0.08302603514703866,0.02070062674891023,0.1726833506479545,0.112730776355465,0.5870276837510721,0.000224030599962732,2.420523410957966e-05,0.06004494455168718,0.0627071883479276,0.6798880178660568,0.3100805541592244,0.617811342118718,0.84279840171814,0.05009308220967389,0.6610994954187135,0.5921937201241007,0.002174562010865121,0.8981768256616506,0.001945476041131487,3.319997169069173e-05,0.01971132750007707,0.1244794686522807,0.2873144722258079,0.7323860466692993,0.07113943400775317,0.2702685091193575,0.01281132495844623,0.0003127508876906083,0.9059408050649425,0.03639816809176272,0.0004281792255491922,0.2879585500841341,5.495476490248281e-06,0.0002193212345146001,3.380420238906238e-06,0.00529261373149765,0.03780412825353194,0.008857525235911571,0.92273907670411,0.008798341937046083,6.111344207142039e-06,0.7417775491710438,0.7069409772484709,0.1022864548683486,0.9188364795036531,0.05362470340547557,0.003721795475560554,0.0677057003815927,0.7867571183860103,0.5106329195195642,0.0005865596805212282,0.7742464447870098,0.05554488865941087,0.005974592116179843,0.9943993628276583,1.014829013900185e-06,0.0001407180016535486,0.000219830078570694,0.07384380343848178,0.853570323397687,0.01833512589160904,0.8425200253580303,0.1485064729923449,0.00173862623692598,0.1955375252350766,0.9580845106417998,0.6388247999253228,4.825422209729068e-06,2.676634767038796e-06,0.1900843108649924,0.3775895810205579,0.02684696707553787,3.789582328574032e-05,0.7003430201936628,0.2514749866220015,1.967276863390857e-05,0.6917399398315528,0.03355549717722242,0.04235830823967331,0.001437222566061993,0.09999424069152053,0.558731011180389,0.6189733357252477,0.0008179406507848359,0.03642235588398079,0.001414430891659252,0.06406045433814891,0.6842665322335355,0.7589125152605171,1.641275083076126e-06,0.3558718694892624,0.003775767129426754,0.0008778312161329185,0.3742911032698469,0.9039735535263234,0.1611550798900337,0.4576950703038906,0.6804334743937054,3.792772158003959e-05,0.007227903218639838,0.09842682510068503,0.1758484596885986,0.03355549717722242,0.1175371170188479,4.084115781821167e-05,0.8714900036831484,0.8799442430489973,0.2658464794173449,0.4722584366965388,0.5291218627774736,0.003353029916231989,0.0162432761386442,0.3688359889502327,0.005054540821692704,0.4394934591816533,0.916178359361523,0.1769471926373819,0.2941231351606834,0.1026970604356443,0.2097279287014937,0.004758794586654644,0.2549137861908243,3.661639638655095e-05,0.1225515908355691,0.004388338281778818,0.0006436910987618957,0.9968127519631974,0.2496338224404132,0.9614698628545661,0.6697948519997983,6.025331334052489e-05,6.570632799135745e-08,0.6909612144390112,0.01754949571846585,0.1321593221281761,0.70558544842823,0.1518036282351554,0.8502767869404804,0.004559340008953423,0.009469346733495806,0.09249757476310387,0.0002995025127030567,0.2488496998762658,0.9413742407396022,0.4856299686553305,0.0004231045297865093,0.3264273527043792,0.209892948122852,0.4315708839790719,0.0454973902339214,0.1456632167784667,0.04080971616425544,0.3494415422342077,0.9462666521674232,0.6239207385311216,0.01976656381565139,0.8803315340772719,0.3567894516612276,0.2065359889340574,0.4288769326558183,0.928639994257329,0.9203355562608775,0.0005951015069993855,0.4386883625418553,0.01754183975888453,0.8945839571098519,7.531887395242697e-05,0.05549355048447589,3.450569850899407e-09,0.4750633881245883,0.465597477607034,0.01706025485702817,0.05833290299907599,0.9341240776372614,0.01578856228781265,0.3143769840317572,0.8004974222929668,0.004261382872355865,0.07942826626729639,0.05777172865416098,0.5949380853459836,0.2714930467082498,0.9131671773663731,0.003389485231006944,0.2915752322400803,9.44822022105287e-09,2.687961116925702e-09,0.2194453904220377,0.07505439098056518,0.720966916644468,0.3306710971123086,0.8155806000438485,0.009812137914794945,0.0801096318704175,0.001496078845018505,0.2771514100736718,0.0891476326817864,0.9243139854427757,0.237016902345304,0.02123119318934296,0.8394108513847029,0.05578955197192532,0.598173077795779,0.7853542962485176,1.379535079674625e-05,0.0001295006722720277,0.4838594388697957,0.04188066384615872,0.178374036330271,0.07370046584406341,0.56028576112992,0.03677199083043199,0.07079654831763545,0.1232483394287995,0.004687626988759054,0.0003242358005489646,0.01191532205727977,0.9995587242417702,0.1279349491440927,0.697038916662684,0.8350611207932529,0.708768878468786,0.01183138894613581,0.3371420714583037,0.0267332013970875,0.5713361082212909,0.1483775434588539,0.2107630520216896,3.235001027451694e-06,0.754437664752362,0.2126429626100635,0.102766096978523,0.002819037196466281,0.07782341949604918,3.136799408704244e-07,0.3613996514391573,0.8598626212162042,0.02062207560822446,0.916286011144946,2.298112223997112e-06,0.0003866404059696816,0.2481970821432652,0.2622831205328005,0.3795016393267657,0.7685965262254677,0.08499987159129851,1.86277064270705e-05,0.08670043917908178,0.3008578072379801,0.5921738104130662,0.02437860396578188,0.06942265476276677,0.002195832834098739,0.005050051492659388,0.0003248590983730282,0.004571615995158484,0.1995414750687735,0.1047382743885361,0.7166630783696201,0.6523355320884507,0.9865358738424717,0.5962165692651199,0.4164784381322686,0.02700289312541369,6.962796343832503e-05,3.138200152690684e-09,0.3661277489949646,0.003083342187918068,0.3447543280791291,0.1387199233881714,0.6777222383531695,0.4844957812995678,0.01417561519307055,0.06917407115034201,0.06287387209665185,0.6558218254648379,0.01603787448479424,0.0001752498300227316,0.01949734789065248,0.03546356895963702,0.5677350168673836,0.002190314427687327,0.04463300034470725,0.00222279976891578,0.3345403809174302,0.2143737904396678,0.01019679158585023,3.864507298909818e-05,0.9441642991323108,0.6408172828474565,0.2261751549067706,0.3599485781567357,1.772410256119997e-05,0.6767604092039171,0.03057730149449842,0.0004244504748844388,0.8005529239463015,0.9314731203242578,0.6525293001712792,0.0001623097648086605,0.337133816905974,0.1722007780569176,0.2212131753774612,0.1759938464600096,0.1624767428207047,0.00590976361176974,0.0002493129347654455,0.002885748574593401,0.4676946208322098,0.05745002150432397,2.169978395886743e-05,0.2393708617674421,0.2203239210558807,0.1034578882210257,0.01726246660212608,0.01900774196590986,0.06527811915846052,0.02635162493787229,0.796929698261429,0.01203733659028786,0.002667313207509777,0.1048420261583337,0.8632995179913744,0.0006294058546189127,0.3438231416730383,0.0006222519027923488,0.09556234708567311,0.004803043477182148,0.05317870565773888,0.3245345406011998,0.8684518700709626,0.1961454993750298,2.934841262467324e-06,0.01145975711400596,0.09042358953139727,0.9667249776203444,0.04437591027790786,0.5648818154021182,0.5883954553435581,5.374047978070353e-07,0.6207071550295801,0.06910385384439685,0.0106611259355938,0.9220474900408804,0.04939622272035551,0.02229324718465724,0.09068791643290566,0.1816531268586731,0.4377222573998539,0.1634118243006534,0.0003721945486218658,0.3247711616240609,0.3978684514236178,0.07145687381553409,0.1589098331732232,0.5248808959194099,3.831443988517216e-07,0.9881960808689301,0.9540390327053675,0.0005869954749934206,0.09249757476310387,0.2887838604236612,3.67941588945557e-05,0.1474921217981386,1.183121581827721e-06,0.2008965249496241,3.867124339666362e-05,0.05833814577252232,0.1326377397962769,0.673398688800559,0.7644592860765984,0.02925730790686186,0.2666143268161918,9.169795527338662e-12,0.3954907223936096,0.8417671936750322,0.006585562439332818,0.872041705460933,0.3072613522745514,0.0229677986343546,0.01982512507101882,3.806227970670147e-06,2.997788172959974e-07,0.3566051426368453,4.372902987796047e-05,0.0104903343688285,0.2027019604493007,0.02051108120959573,2.164427589813418e-07,0.5879157329304635,0.2838281718726425,0.1722709537604832,0.1340829687365651,0.05076991352991524,0.5276540197315357,0.4722584366965388,0.1853411134664369,0.04808762744273664,0.7274188467156297,0.6169686680500346,0.8327670310797102,6.277818998329348e-10,0.6421127216926329,2.3101864458367e-13,9.340514126845577e-06,0.0349721368091726,0.01081136712201511,0.615726001751347,0.2577145436742577,0.583489161439248,0.1284738441299243,0.02047093078864699,0.4535257949300478,0.472267809625374,0.892216996241968,0.08642598872230169,0.01448020358930482,0.03245961503683582,0.002597586402533852,0.001130527304841546,0.2090193487505061,0.8438001595498383,9.47029797317708e-05,0.1316145588998991,0.5840703059031145,0.0001759647145071584,0.1975908536574611,0.0002154553980320062,0.0009112455380779819,0.4787128861190729,0.1088978332043097,0.891131492131317,0.9106587468460082,0.009530275517164259,0.1648011722713242,0.001613100803203325,0.1532955826417857,0.2188695135251888,0.9398932889846108,0.5081963102944531,0.1286841278478121,0.04441421756087027,0.9793259069237565,0.001305869148310978,0.5318478153243033,0.08613360589124897,0.001089016349904905,0.05263213473818302,0.5930744002589982,0.008575464146450168,0.01094668927289329,0.04119680756027107,0.1664097786045729,0.6588857542065193,0.5085385685433872,3.889659713050486e-05,0.3349756542107239,0.3197867835433836,0.06652341308710344,0.313967343188005,7.438108648094837e-08,0.02689318719701441,0.1451966331661736,0.009835561563995215,0.07934710345355404,0.6906543089424633,0.006964988900611057,0.04164774361845668,0.1388538499480837,0.09537090805836869,0.7205879598368389,1.972771732699865e-05,0.001403758108530266,0.2953856886417403,1.453162676698367e-05,0.001128220784887271,0.0003071024590185029,1.008581707827744e-05,0.9816133928300524,0.01154207792767153,0.01317088139906965,0.9137989299591078,0.2195801491294422,0.006221423128389049,0.146601647236161,0.0236458323638729,1.124383541307976e-05,0.001187359267352421,0.01252340179814772,0.005038097616720134,0.1827392873338616,0.2121094018358642,0.8580608703686767,0.3822437158387152,0.02486845145559478,0.2517968353525634,0.661995536050469,0.8667863331920951,0.615359909322637,0.01829060761736504,1.197486954082552e-06,0.09956372612435817,0.01069684481915701,0.01872387482830276,0.1985733196405308,0.06846548994901847,0.000137558014595522,0.08955579541609134,0.2341252621090457,0.07211509036241204,0.6165963165581203,7.221038555477181e-05,0.05707331258294381,0.03176419123158525,0.2675953985814183,0.2691280006603192,0.9205095066495995,0.0001166668372881744,0.0001478350082516555,0.001195839380927921,0.8628110277216791,0.0015314273924847,0.1006740082834399,0.024188922289476,0.004942753424310664,0.7745327958319056,0.5743806252423945,0.7765124645128426,0.7639907820811406,0.2112700165735877,0.001022160605274577,0.6262822684128501,3.349811951241036e-07,0.543872487343095,0.3989207214712465,0.2933486767500329,0.04403764934334176,0.6354152256612294,0.2981710807168712,0.2795761266315284,0.05645629408325904,0.0008680442411822454,0.4495143027683829,0.02303085719552698,0.4930074029207021,0.0007371044300163378,0.5962050333913168,0.9890035842405105,0.4548569607119367,0.6164231694086904,0.426978514767022,0.0002063510584245812,0.0007152459492081425,0.1531354615471521,0.053737568846341,0.3107547020609976,5.249800732817813e-06,0.003881328432631003,0.05325263615656073,0.3196123185561531,0.5068486097620415,0.2531389176585562,0.0188215490867183,0.4478461990219065,2.59080314045886e-08,0.7909339949579981,0.3599485781567357,0.000195134369798091,0.1539999096082001,0.217284987570413,0.489917907372616,0.8938442570527766,2.646759036418613e-05,0.09008953428406867,1.80597478245666e-05,0.0004787636856555726,0.004630106577608729,0.1381015030246562,0.003839865548640919,0.2940361877944015,0.04102606527433518,0.01807234328890836,0.1587782140793634,0.02538022835489618,0.003114876396958284,0.9465753333513214,0.4357644668171088,0.8371178248661907,0.8689196485190717,1.228910421622095e-08,0.04242066949269406,0.943798463842377,1.300172301061273e-05,0.0053483021744998,0.1058446451992179,0.6649787284657631,0.6001234084644145,0.0008889243307466199,0.2842293907203442,0.4412150341646003,0.893070278442109,0.5910696145961809,0.7797782609077046,0.7553862260346776,0.401384803995547,0.9050170909599303,0.8216413003510613,0.9864006330825941,0.1326974620447063,0.004388851502922462,0.8306327339327275,0.3370547390236703,0.8200478747596079,0.0002208751928170139,0.0002063510584245812,0.02684696707553787,0.84409763165856,0.9318824504050707,0.7069409772484709,0.0003418636254690881,0.005160519702512472,0.1734834882980639,0.03242861348977193,0.0008896931306782727,0.3275706569446336,0.9750869035920962,0.1166435804158289,0.5369476113492094,0.001917980804477064,0.01255460922524552,0.750735814442648,0.01616177400169869,1.177545294403017e-05,0.2985524171489234,0.2418935860915506,0.2791233732030577,0.2091949373460015,0.3475223102317266,0.0003716348929678494,0.7236413834923136,0.1494792908706646,0.1799299572708256,0.003029032682167904,0.5618414130523139,6.376679294335727e-05,1.143940596940014e-05,0.2938654199501602,0.7293768159436351,0.1186088357949926,0.6847388406509608,0.7641975331731772,0.2765838603292687,0.5890457804345194,0.6919906585879835,0.6203554101174856,0.01729967028031711,1.595483684370515e-05,0.0007704882036354197,3.037260039107859e-06,5.479278731001965e-05,0.5601434880450308,0.003358114110926516,0.03497741552024178,0.9078770692133724,0.006079680795584183,0.1782313050521754,1.94962615532982e-06,0.8616642987357883,0.03047144835434843,0.198304923515106,0.1479033020752943,0.004057909245011869,0.7715819630321329,1.148202524308098e-07,0.5869193208712619,0.4283509649202249,0.01719562423499367,0.02304223239740988,0.0980659634503469,0.2204956534004528,0.583489161439248,0.7692938157637897,0.009682912137875144,0.636377457219619,0.9462666521674232,0.2865549831025819,0.5910696145961809,0.4271612502601277,0.020153794198501,0.6900283914044378,0.2700270618679569,0.3866430466365954,0.02273117285354411,0.5135484133306357,0.1080494894779778,0.08387132600651181,0.1169578596391492,0.0005422121538120588,0.6817179267201351,0.07001554523736253,0.3517327697745789,1.001080568231463e-07,0.322669518284225,0.2137568158783803,0.6198146201822144,0.01532443849377301,0.0144506273449295,0.1508368543080564,0.02051708945913325,0.2505430370448207,0.6734434660698077,0.7404193472768029,0.1739115894593544,0.0003744441265927863,0.3568460824579699,3.944397141280959e-06,0.08887304754576812,0.7725056371449817,0.9004855408424369,0.01420102773129018,0.4423851932422832,0.6082901876385024,0.8332081510916829,0.139795889886069,0.638465524428477,0.7680962145636024,0.2992897341518017,0.005345685481204383,0.02265289328466056,0.7801092185523277,0.00287359654304257,0.002446746942892272,0.0001166313683451042,0.8137420783296719,0.1216699414909406,0.720500996378955,0.03825796676602171,0.006395071964426731,0.766214842350561,0.7685965262254677,2.296187441395138e-05,0.4249955328373319,0.2873879937947582,0.2011705623051645,0.4176899663973457,0.1859703242323357,0.05669164337175185,0.0004009792050755584,0.5843480428333477,0.009465132766887763,0.002012794647176453,0.04042821228898063,0.3376273343482369,0.5168386005500162,0.04188197558289317,0.9180101672337828,0.3089697395482341,2.615270620790761e-13,0.6290888415512228,0.0006973036759653961,0.1090827340101798,0.0003755518013092251,0.1198209876927328,0.7570514425346156,0.09899232418080722,0.3825305465998745,0.09817432033028588,0.7300322222177172,8.545145074798764e-21,0.03702087027192602,0.0004606189405160346,0.06791010278992007,0.002184649097437115,0.1513623855466487,0.626278339905781,0.2299934526171774,4.040958488012496e-06,0.1248605148206163,0.004974337261487276,0.0004102631728919244,0.3502025937301083,0.03978833444934349,0.2871283990977446,0.7532240459234625,0.05033772648458525,0.6111004396243735,0.1581829948254229,0.07047950589153276,2.345523390989491e-06,0.5441267241231195,2.09154139800042e-05,9.491885297668771e-06,0.9761142040787655,0.1691210780255656,0.8183346069199274,0.2242358383354084,5.788950962179733e-06,0.2203894342735847,0.739283885311816,0.7070733071209822,0.6326096300855015,0.4144754537606743,0.5661961438458404,0.1454721823682632,0.2816730004344655,0.01708100537267731,0.185980378070595,0.01239413337559861,0.06868405210601589,0.8846650052665372,0.02062207560822446,0.443781506188003,0.2318560076439296,0.4103538420923147,0.03826733218896375,0.3260875443039085,0.891131492131317,0.001405012588313763,0.02986532857498803,0.02831257498635087,0.4362673239897327,0.4881738409708696,0.7535787764301751,0.06177735620468854,0.7580898564187131,0.3626479339782239,0.008790906517980561,0.2598093004874898,0.02293994255632722,0.3107268500888526,0.9703616037784075,0.09192666207683665,0.04391062989895161,0.9285995070563278,0.05356737028089055,0.9082347228307805,0.6198146201822144,0.8391807376617009,0.8381705512621298,0.0020135154816408,0.828345641666084,0.01189778356605664,0.06255954945607742,0.2003036468604838,0.2456423513575863,0.08613360589124897,0.02242955281345583,0.1033562626819722,0.4071606843940209,0.1381270771605892,0.07515578715229576,0.9966565151720415,0.3254139546971313,0.2711580328389318,0.8929640239920813,0.8539762813970959,0.3702009367886944,0.131058460247219,0.9772277390631341,3.537823236460175e-06,0.2292160063115338,0.1871095136358978,0.002906672057485874,0.004449510393386185,0.353919697544578,0.1127993027758567,0.001457595527531872,0.7623046174376032,1.735117476480303e-07,0.0001280919746897234,0.9162944804220023,0.03484820522092563,0.2319659879292893,0.002800838760699741,0.01600043390527272,0.0002273747530624452,0.01701256097183552,0.5909571021914438,1.415841413182945e-05,0.8656791344633677,0.9596248113465647,0.4572774730832227,0.2146344237093845,0.5343453217097542,0.01033876410734699,0.02142091013174422,0.001188756786685886,0.2999483558450881,0.0009851898336398473,7.040631211422363e-08,0.003716868974278771,0.02240167407635482,8.291189864660087e-06,0.6129158869642772,4.701664129219104e-06,0.0399848012240607,0.9629000248698629,0.6209619401816762,0.1444299157183492,0.1398228499987772,0.003127836572512393,0.0002698210779690124,0.3778609051326162,0.01238039299062114,1.006736377666989e-05,0.005233825517076026,0.0003520062902870756,4.581882276246792e-08,0.2457781192026283,0.2648139911344272,0.5742519903763846,9.502659215982844e-05,0.05381719188289224,0.0002582455008457752,0.1226088517448439,0.5604344608820743,0.1008684330478301,0.03398007909537574,0.2778801261509926,0.008791341198517742,0.514430673945831,0.01204306061736569,0.8919277655371299,0.3593382070929777,0.03259189191893334,0.005451610658629387,0.006753264838231886,0.1206418438675311,0.06912362795252523,0.0006748507876928551,4.134869569379778e-05,0.4094267303201716,0.1321561583377368,0.1644725329877531,0.7841610591431083,0.6415722344877193,0.003324157460936145,0.3303996390924933,0.147983884522616,1.339299505268283e-06,0.2080727240190052,0.1328808311531003,0.3319168190581457,0.0007365014873588821,0.1193259841743207,0.0531987153845376,0.005379047753630566,2.475534411318094e-06,0.4681488411889431,0.8103612630955417,3.681556405541286e-05,0.7684942532846267,0.6444873276633761,0.3563907188330692,1.85222815814763e-05,0.735423134191913,0.7722388421342522,0.126958258769161,0.04495751688302686,0.01033383947667284,0.1336744230933964,0.9137989299591078,1.643591480563512e-05,0.004388851502922462,0.4221877288452583,0.1180724368964619,0.6769842782332771,0.02228107352325983,0.6408362028484291,0.01783531242896416,0.6747199787495978,0.01109456100444159,0.01591934152121418,0.6092840797594876,0.9366002428525789,0.0007305101472009756,0.4203577889883934,0.3413620387258703,0.04284542794359893,0.118761966452148,0.5380874690572496,0.2195801491294422,0.2175345629182861,0.8214358870892194,0.00494540435240398,4.602546150406022e-06,0.0403850931145977,0.7909971442920076,0.05702569723268602,0.0001270290914355896,0.0008374564799913071,0.05967213099560646,0.3376845566851149,0.5460568103294948,0.6290888415512228,0.6288567108153152,0.0003489206175389219,0.8636637488763087,0.002009121951830954,6.669773663211107e-06,0.1058832461809348,0.004277194471377205,0.005838877077213073,0.7773514086662722,0.001930569326794137,0.7065790436814694,0.2689436839089307,0.08589938362499987,1.28417549721358e-06,0.08563266346601357,0.0006302626388839385,0.001513694063890851,0.02224544112606938,0.000284279300153699,0.002828853537272635,0.05187929082270348,8.164067989795679e-07,0.3294198849583681,0.1388709840335685,0.3776244937434788,0.2358095840220577,0.630587443392949,0.001524116776893743,0.02266826522458685,0.02674016566039333,0.001389312825914711,0.2198213278710222,0.0003547451746183731,0.5830412117353948,0.5166994489210504,0.8972735199531928,0.406505234165221,0.05360249000094939,0.0001849048909055422,0.2378403408741663,0.4098882077413215,0.4070972001416223,0.0001806780307939371,0.229073848294496,0.02123119318934296,0.184523716210199,0.0001248712531458483,0.3946826669058223,0.06845632523502081,0.1714923070755158,2.067774284273427e-05,0.5949380853459836,0.6344782389393422,3.675596725371129e-05,0.8448825728047383,0.8944966158104456,0.08168742116429327,0.3264513171104193,0.7828053743009487,0.4002942972191905,1.300172301061273e-05,1.7483883080325e-08,0.1193911624832344,0.1659607789619248,0.2615626304209668,0.7065790436814694,0.7939184087758118,0.1472447516725917,0.001129151141430648,0.5510495969831998,0.237016902345304,0.2550237083084927,0.01947823769648487,0.004591501043457177,0.001007406904531835,0.6785722661774016,0.05171168726172844,0.74160342967451,3.638648073223419e-06,0.6404009288654757,0.4541750635176672,0.8714900036831484,0.4864366409855672,0.7967780720433044,0.4831433565384818,5.487188332146683e-05,0.02538256052284562,0.942775902031585,0.1554798183173212,0.4678378048116661,0.0005425988378734218,0.0005406958221652452,0.0006119753452850894,0.2070323138899433,0.3143769840317572,0.0006529829982800243,0.1133105596002472,0.02057911482263017,0.454504246501075,0.002824875002648409,0.001287797786656862,0.003814681472665207,0.4368443455856744,0.001686413559184788,0.002629597216918795,0.002073226461249527,0.002085046575353607,2.200744156495598e-05,5.38638858787041e-07,0.00112556133479272,0.1141567309066467,8.122682917881586e-05,0.5950497110426419,0.03087945221076532,0.09913037331005423,0.000623562426360559,0.1362997318939603,0.02497109169653103,0.0282811578158811,5.337474465085381e-05,1.871556273754242e-07,6.000868169833875e-10,0.01142763190987285,2.681785656989605e-25,0.06618578361263845,5.325605443276391e-07,0.9871167703679132,0.0354736409104148,0.6940732534165956,0.6688169587245663,0.08552933387550643,0.8475762831286417,0.4478748750518383,0.8653299475612293,0.1044014605298139,1.032936761097114e-05,0.01297800408557377,0.0002012269238975522,0.1230718227938799,0.103923514537761,0.04580611367091428,5.17089448004061e-06,0.339167866466386,1.115563089656359e-06,0.3752628094642079,0.01466526313597923,0.1390730408053621,0.03169028107014785,0.4478461990219065,0.6773855046530396,0.9815661993228887,0.2274988524155691,0.2322811550484534,0.3702009367886944,0.2146178484773234,0.02126491314168662,0.5043132366063339,0.443781506188003,0.1085969712045524,0.1007900666992902,0.009569206768556928,0.0005664355194922976,0.001405012588313763,0.04966547232259115,0.0008138939434445339,0.4085907343048882,0.2106874625765264,0.9607914407058263,0.002102036080249041,0.002953052735498981,0.84279840171814,0.1460319156012926,0.8350611207932529,0.0001129890794023306,0.0005335012803940638,0.0004205396521477203,0.0769990826252107,0.0004448809421574444,4.214452979808252e-05,0.7956631647387727,0.03006858441015182,0.1725796953569214,0.1841435494257989,1.377126045591202e-08,0.1705192621317165,0.485200447378414,0.0009112455380779819,0.4373196337693673,0.07047950589153276,1.463796641268269e-05,0.8843249868668023,0.01995062885320131,0.1648078475422853,1.926818211905558e-09,0.5728484855246055,0.009681246405027193,0.1188623821027018,0.4128652427576983,3.436282171953506e-09,0.06161507686186028,0.1988806220638461,0.04031262553750017,0.8827622376909376,0.01824549933615651,0.3365975883509396,6.835110128190038e-05,0.9015245125758632,0.7898399195306983,0.7609494344372136,0.8042254067488582,0.7321897479249756,0.04906012283607965,0.0489294400673741,0.2853360623453374,2.745329548348951e-07,0.005449603729898886,0.001381071646704945,9.103047396958655e-08,0.0001468727386559172,0.002450823481247073,3.191311878725839e-05,0.002903680100076426,0.6951468359036616,6.74849474219571e-06,0.1369297339078819,0.03187965491103798,2.299375507721054e-05,0.5021980235554665,0.7685965262254677,0.3570977794050131,0.0006711225281884849,0.7815088741011312,0.4290909753325429,0.8404135268693025,0.6424675133823472,0.0006586767997341662,0.002895725669242377,0.2561343483063617,5.206051530645948e-05,3.450569850899407e-09,0.03385576117957812,4.521390664380563e-05,0.0500823952275873,1.945846808540663e-05,0.5214818270579188,0.4263548690975392,0.1127993027758567,0.0354736409104148,0.1602918555308364,0.8806126381781566,0.06180144681502703,0.02224544112606938,0.5062596146873487,0.4410646813830713,1.706912292142524e-05,0.0053483021744998,0.5087640325179282,0.741853948048094,0.6677737513523793,0.6859997115327724,0.3369238976173416,0.0009946246282929382,0.378912492605614,0.6533129844598015,0.006666324360077331,0.001417708759507125,5.616479144108348e-06,0.5130919144688236,0.09337468790871653,0.1467341141190251,0.9834493731662909,0.4103223072872096,0.1624767428207047,0.005828072730308436,0.0317903541934815,0.02661674840552994,0.03031394067634845,0.6649292228424223,0.5471258586982676,6.73634735882661e-06,0.02384839940647603,0.551157586676237,0.007905740055721852,2.721919518097828e-06,0.9875742549377977,0.001539236712265653,0.149902915904909,0.9667249776203444,0.5037330436573185,0.01133809457984568,0.3349135614156923,0.2702257439167583,0.008037509169376563,0.9901999461317573,0.001381071646704945,0.0002516249478171597,0.8103612630955417,0.0001204586599828806,0.07926539853871427,0.005050051492659388,0.01982061184537801,0.1148960249533215,0.6095404457380045,0.2711580328389318,0.5545180362103125,0.0568965244907855,0.9801915768125048,0.2613689302714907,0.0007783230027629686,0.05325263615656073,0.005485141575096107,0.6600779944934894,0.5438413411476832,0.1425761162217739,0.9954068862833675,0.66151330970446,0.09884755052556152,0.630901211093713,0.1086887304587986,0.05327173110258927,0.008925592629280027,0.4622490841152105,0.03221967143435773,0.01522509074622407,6.61848633669552e-05,0.03891613853787958,0.4575995453073931,0.0001576011111703614,0.2953678008432158,0.279390561007644,0.00902485559469886,0.5037330436573185,0.0006729636019793502,0.463225132692161,2.609168353792877e-05,0.6734434660698077,0.0001957935844245223,0.960374115840723,1.2656129351496e-05,0.0001270290914355896,1.511494640143036e-06,0.0677057003815927,2.244512459023439e-05,0.05548797995681204,0.986360766665048,0.6169686680500346,0.8914911370690122,0.193084642842388,0.8846650052665372,0.03354652497167926,0.6123995074860096,0.4281192557411718,0.7360360591809699,0.8231007849825646,0.6154086925051814,0.5579155582655022,1.572345909389664e-10,0.05885807621887162,0.458909645863772,8.469190791960077e-06,0.8035012017909385,0.02753987657654097,0.7262184188444725,1.371426512794755e-05,0.6971135165920305,0.008669224765837853,0.01780908077830546,0.07782854155989191,0.2728758979571717,0.3582677193456088,0.01882603029988053,7.361098534774483e-06,0.03509883801895759,0.7994991829061829,0.02617369934564612,0.000949233120066489,0.4896716821335315,0.2557041673209182,0.9326677445173869,0.2981891095217985,0.8077078715567256,8.511736669402398e-09,0.04341802301559013,0.8527482648903019,0.2068325244885215,0.5190533832822749,0.2915101161002061,0.2915633155357546,0.9460870557346376,0.01959072050252133,0.5052133084077898,0.0005553102331408659,0.0009820163478020997,5.17089448004061e-06,0.2164628936381932,0.0005059026267642199,0.2489224099611058,1.207704815889055e-06,0.0005475614526568329,0.1342646405830685,0.1330577728423328,0.0001344914444730803,0.7084171399247994,0.07087390937657154,0.3081560869492858,0.0004045844443583071,0.9053986336677005,0.2248347012453093,0.6571992999212716,0.6408362028484291,0.5263782881041098,0.6141350582696578,0.25823325810093,0.05736143296361849,0.06864399778817505,0.06272170380928502,0.002484355701312278,0.7064748715671327,0.3059277181775069,8.204974566986465e-07,0.8137420783296719,0.3984356330344446,0.0124519004396997,5.187731131272057e-06,2.632426997480512e-05,0.1005034275280085,0.1860474308227127,0.406505234165221,0.229073848294496,0.02824092293362465,0.01381185268998539,0.001599346208333772,0.03516497326557282,0.3571505912406167,0.1639548356355547,0.003610422479299045,0.01408410161660122,2.075515395380065e-07,0.7940634071248093,0.000623562426360559,0.4249955328373319,0.1772355471272335,0.08686385365340812,1.096123695086537e-06,1.643670452943298e-05,0.9117619935249506,0.02094286447036438,0.1958396661959249,0.709080734269152,0.9844489058736869,0.0008498623967378347,1.322304881339598e-05,0.4803278632788074,0.6195511128666195,1.869444640849602e-05,0.004898814855587213,0.3474980444048233,0.7112758362984893,0.1453819937866792,0.05205690007224022,0.1714802413730226,0.3781863458248805,0.0008219229249300773,0.1492085951416378,0.2123305157609463,0.006055260536876017,0.1512682193317474,0.1092330319922355,0.04229655895393186,0.6078013646973084,0.03335019657241991,0.6345206647050011,0.168337489243471,0.145853287970975,0.2087340898778791,0.001445735740554856,0.9337651105246443,0.3294016670047386,0.001682930460074283,0.8910399672511812,0.1128917283669721,0.497723891683111,4.344706116075848e-16,0.06180144681502703,0.1110491361018899,0.0253244866437492,0.007905132436802912,0.6768386406242779,0.954938950227045,0.3726342303985191,0.7348331461846602,0.1050320191721007,0.2941709282165308,0.9164119256636791,0.4319490377392304,0.01803401426875245,0.4930074029207021,0.2837071475022145,0.6931577258779669,0.02949255737344447,0.336113067407865,0.06669394445515853,0.132537843916998,0.2571978987015384,0.3978684514236178,0.01347101995606912,0.005379047753630566,0.4170916716463355,0.9943993628276583,0.02845671013859419,0.9524919743482566,0.002441032995098976,0.7024750835677513,0.3867904718011927,0.01371298698717878,0.08392049079530861,0.05649275334681458,0.8846604011080774,0.2194772254441836,0.9567225500781401,0.002343599230768197,0.04816207855441147,0.06190813615662838,0.6043412558238452,0.6776277229756986,0.002180035899251738,0.01893604512646775,0.0003996176947793556,0.7333059055364348,0.002399242089242001,0.9769524554065943,0.1162192904073535,0.233973979414663,0.0008166873099222886,0.3475950953317124,0.008518291758332769,0.4419521434325333,0.01846930312063246,0.2590984468989642,0.01740476882965247,0.06666735583661894,0.01239413337559861,0.001824612607663992,0.01301921031036496,0.8497249732880033,0.157720771253588,0.0009253244798968724,0.003149592805951121,0.2656697162915225,0.9368770779396367,0.02568699475452253,1.303415417166638e-05,0.6443755133032923,0.05399988663059609,0.1002101794283396,0.1797697610722665,0.5689483824291779,0.00625301049901761,0.0002033434312287559,0.4763048455321773,0.566255952379058,5.519853603148214e-05,0.8752148532844932,0.001487359325696049,0.03127425508102865,0.4907494903224542,0.8294954916541089,1.226099028593047e-05,0.04722213710508816,0.110422123293955,0.1827392873338616,0.3611067928513189,0.302288587936429,0.3381872773470462,0.580092546011696,0.2662953639234598,0.05989251633115842,0.7858526226803495,2.302621935135765e-06,0.0580439924062883,0.5139746536968679,8.293902611510825e-31,0.9894953227737648,0.9852667686209932,0.9034015663783298,0.7524022264712409,0.0197797212139446,0.02356535450158217,0.3853030015737532,0.5512752545767324,0.03932398308904385,0.006270307565059485,0.8252125844347429,0.3781863458248805,0.4489404593874776,0.5648818154021182,0.6564839853632175,0.01585100917439998,0.8446908723982686,4.553928851143984e-06,0.5774748124570301,0.02672665463644206,0.4164508718191328,0.8724625576665805,0.9208862415278946,0.005878059073209253,4.899463450389374e-05,0.02504970235160674,0.2025943748783961,9.077093422216568e-10,0.2829461048253314,0.720110720634911,0.1454721823682632,0.002504828849136673,0.3612732572676579,0.01578856228781265,0.07293943327669528,0.2125554186374103,0.2004505626959373,0.05685535609014433,0.002379675769618932,0.5689186646957942,0.6388776417423204,0.4861592061243277,0.2439538687107402,4.316641658779235e-05,0.08441340471304684,0.02146775555290754,0.9243080587858583,0.0007351347795966552,0.0007951533707232794,0.01496065690132763,0.03294148818846861,0.9489234071730729,0.4240018679798216,0.08152630620096328,4.045086200429624e-09,5.866015136019709e-05,0.0007478971211273831,0.7809205939192193,0.09369637347753097,0.8946744598715128,0.02360272895800903,0.003680093764058697,0.02768270339164583,0.7415249326363885,0.1819280533319041,0.3112887360437683,0.1196180270374444,0.0004369077103797668,0.08191088965262962,0.004681268986739969,0.2686248466849004,0.8725309857393673,0.8828746846554543,0.5728484855246055,0.005131738936278674,0.06600256530813604,0.2479724464726876,0.01431081489813677,0.0859762709078752,0.313967343188005,0.1547190551727448,0.7178954400067372,0.8724625576665805,1.157592811800505e-06,0.8580608703686767,0.008609736693208929,0.826176715644475,0.4564966048012675,0.4941851483482638,0.3604677826090418,0.05545675587079055,3.394485994565736e-05,0.3563907188330692,0.9878613443108506,0.05560486535867539,2.420914857868457e-05,0.0005154808276657573,0.04590635962209652,0.004159708106491614,0.5139746536968679,0.01512236111814986,0.3080793735661395,0.9612622991963727,0.8523470305995665,0.9732137192683574,0.001429122406972338,0.09254635138857564,0.009530266258106374,0.01790005384343275,0.6955968312277975,0.7918914374082865,0.1745117027815288,0.5896926863515476,0.8460126507081173,5.926751633634038e-08,0.0354736409104148,0.072749399690999,0.01849337127172294,0.04242066949269406,0.5022601725942661,0.324973068944282,0.0020135154816408,0.2485904484454067,0.8550153786967879,0.394465400224755,0.2438366008161,0.0002118097838687881,0.3058174281087933,0.08825045732511583,0.3346030135960505,1.15405318143139e-07,0.07959670840223418,7.255725057171722e-06,0.1389284925147256,0.07859746011739967,0.7858526226803495,0.0001206564045529898,0.7704497752876311,0.07199876805451973,0.1586945434159023,0.5033132224789529,1.399983686084429e-07,0.09210912159736379,0.6881991772283675,0.002123595134078497,0.02906762191169878,0.2363430887385974,0.02189876744850588,0.004129727771712046,0.4167009477444967,0.0206360039075175,0.8227056143931116,0.2915752322400803,0.002948671172715953,0.9599131545393762,0.3850476659742846,0.1958250370169144,0.2701028111664951,0.5092315255628653,0.6821793180102554,0.01503774964981457,0.8333925178856707,0.001486903990887094,0.04758547609782619,0.0176909400794097,0.1193911624832344,0.2097279287014937,0.3988390077615484,1.522662851072767e-05,0.01648428716378026,0.6698901210493172,0.02895317710691626,0.01595728306734644,0.9547850088301953,0.06818026995287874,0.5019260980441818,4.427924506362763e-05,0.00183596112569291,0.01608169434084378,0.2947010457481145,0.200183075139364,0.08834420177104232,0.01625093356011097,0.001925268270992423,0.4550636356607689,0.22497442123457,0.3535242370533792,0.04592044264058315,9.340514126845577e-06,0.586715530622847,0.2205835409417289,0.01807425282517025,0.9516919250380401,0.4243696619766324,0.0004068153545563113,1.367096665521633e-05,0.7768601985829617,0.1177686119654706,0.479062291161756,0.1134281323008961,0.7012804476502231,0.2873558483599227,0.01561944204805135,0.9852667686209932,0.1483455289705558,0.2429668801246493,0.0001084535428010268,0.502759765835985,0.1696794863315639,0.002660746822002596,0.4217984643650703,0.4761571581330544,0.9577098476410134,0.0176554614006167,0.637996348711964,0.009655795566902162,0.0008196482424604923,0.8298303192282441,5.215615040317866e-06,0.0003392393140707998,2.253617681881456e-05,0.005326384059785618,0.008188225847442906,0.02118193397872083,0.03221246272205317,0.01137860027491779,0.02489107191931415,1.139944284424414e-07,0.05055910145188001,0.003030732091944837,0.008952783410479899,0.1687948595689453,0.8846650052665372,0.9383429046723291,0.7511198268814243,0.9653345270751076,0.007114509677318201,0.8508965573142021,0.5627853082050208,0.7531923268585513,0.8518865819123895,0.3206955236024887,0.1768300615298567,0.05312641137581793,0.002781389490672651,0.2457986667242376,0.1644419118977993,0.1951016009088399,0.002667313207509777,0.1017628605776005,0.6173502085067361,0.7848119543503588,0.9222566796396091,0.4101225675215726,0.1193911624832344,0.06725155631732489,0.2772354358182025,0.5284294346010141,0.2839387569862209,0.9732694137993985,0.01420102773129018,1.925272906858639e-05,0.2500510674211911,0.2786454162706927,0.1074204118707584,0.3642529880741628,0.05347598566893114,1.600063557778173e-05,4.611009386006121e-06,0.01046304090426561,0.8416070842721765,0.4722584366965388,0.002242140049315061,0.8875093449417368,0.2485283607820997,0.04739366020756192,0.4909599591015267,0.7352293498029532,0.02715091365551319,0.339453336536689,0.003162859109683819,0.4155943084013178,0.7664827833276338,0.1999989496916143,0.02022019337225756,0.7828053743009487,0.3569140531262214,0.8118341667340282,0.001289279921065855,0.8619903446321425,0.000370349016366549,0.0713303209000751,0.1702413832885345,2.65160925597619e-05,0.108648525816442,0.6293567068822725,0.5139746536968679,0.1013718710992879,0.001062819257688872,0.3991827976505936,0.9894953227737648,0.6582392784462318,0.03207596172209569,0.4046234114889801,0.2009674732362589,0.2959309362635701,2.555566108254817e-06,0.9256740228756268,2.11128419781252e-06,0.4576950703038906,0.339451575770031,0.06852745740816372,0.0002711783733218243,0.373561564939967,9.890408699859639e-06,0.6699408684527994,0.3351870013888739,0.0513633634099382,0.9283305777772706,0.3939346099722779,0.5400539436492904,0.9152393490467514,0.0001760135966931266,0.6986542641793809,0.4841166748578061,0.2616611723118321,0.2612135671707228,0.3068821694857376,0.7392254440177598,0.3385436847582683,0.01503919977092305,0.0001163824534244909,0.5763593235047423,0.2823969905880634,0.0001765160465263163,0.1344131283104411,0.6554662103085966,0.2452876226373029,0.9524919743482566,0.4087082349669418,0.2042183304769264,0.6781009381608487,0.01278081599859078,1.644902213046359e-13,0.1708854403783627,0.9996962035915474,0.0002132167749679612,0.0909547849611352,0.5573716017944765,0.8044392888045825,0.9738625973363441,0.7061023607995531,0.5211635238666167,0.2071193889225955,0.874664028679611,0.03070011674394725,4.820372405035103e-13,0.06494491564038493,0.789146530985791,0.3869684868200712,0.8137420783296719,0.9666269165126723,0.01790005384343275,0.001517262422757541,0.2837071475022145,0.08613360589124897,0.6240630752870969,0.2476131022387012,0.3119733640530555,0.02250037740049012,0.9730484637109545,0.007031174375238802,0.3920758818113491,0.8094858348044712,3.366155745782559e-13,0.3475223102317266,0.05192798259793301,0.004824670604089095,0.5927354395374013,0.4046234114889801,0.09595156123055587,0.005227284585905667,0.4146755819412491,0.3761062153511685,0.09018893639571206,0.02508388611835065,0.0002489142914264385,0.03856238827770338,0.08872201504892638,0.3978684514236178,0.2347151833867424,0.01068979154910388,0.4477282678618563,0.0004305784936271511,0.5521251202699506,0.01988745043815919,0.05220899541337683,0.5248808959194099,0.06149008336255185,0.8876333576935422,0.6579847436383954,0.0632313464865127,0.3558718694892624,0.1470995247601986,0.0001765160465263163,0.04247579390473809,5.616479144108348e-06,0.007598296379763065,0.9362235521451817,0.000469949129450353,0.01452112677608642,0.003400228006503111,0.373856780948723,0.3186352016541029,3.625618985982334e-08,0.6589280056993896,0.003107797113722167,0.7901777693374957,0.03754440834812497,0.2209920501684925,0.0001222388058025412,0.001741136805842823,0.05264269899068515,0.03146169597889585,0.9655277314578948,0.0641037090438584,0.1676411384032696,0.6788264507371096,0.2527312456675071,0.0247020157496858,0.8091189256552372,0.7579533212937608,0.3629937620115812,0.4100187374165007,0.4468060751460923,0.872302872471432,0.4731452843170634,0.7649538817838109,0.2294426209680709,0.010210191875781,0.001521016946047931,0.8453117852263151,0.7967780720433044,0.3332533381388386,0.2400652061770047,0.8273721113143275,0.09271640176378557,0.03353857560997232,0.7486148322933188,1.697173618258846e-08,0.008920494819483557,0.6701635421875277,0.1784877591627087,0.8564610357247064,2.143551184580172e-06,0.2025316412596815,0.835194083369557,4.319872425620813e-06,0.04262481261138833,0.01339789285226338,1.2656129351496e-05,0.02179732608178122,0.004433700169145061,0.0008198699102194628,0.9350905013127996,0.3740230251743437,2.317973804069883e-06,0.566255952379058,0.5896856628209202,0.4824068951707363,0.001749901210715591,0.09156711988825053,0.6098069886245957,0.002317268231180235,0.9520134533604213,0.7787138678519878,0.6562002579987023,8.866305209682128e-05,0.8726395261002919,0.3028750888216661,0.01156107528603913,0.01113762398099601,0.08563266346601357,0.6167427945919683,0.006877307241682583,0.9724916132803773,0.2632344958028394,0.04529366925092785,0.7657410577290319,0.9340646862996124,0.005947442410498673,0.0007015784054660294,0.6590773652780622,0.7493653782030736,0.003590284206427541,0.1095594723302204,0.4807374071541434,0.001682488878649037,0.001732812111202001,0.2474932975299636,0.0003416924904558163,9.015983660972744e-08,0.8981768256616506,0.01585100917439998,0.02008455754369164,0.2100986853397138,0.04046941330144945,0.08199129239422512,0.002602394632546841,0.1423220912759735,0.9340646862996124,0.06463215933264727,0.08311732220243676,0.5903216018403359,0.0001348910688780913,0.02354117814108236,0.01072922769351805,0.001022064378687996,0.09971699151894915,0.0002256046485991235,0.1142561768309885,0.003810773744319425,0.0006899532730475892,0.7641975331731772,0.3383737170467864,0.4375867726236236,0.3200305824917961,0.5175629388112672,0.6551007954439151,0.0002597648778609288,0.03341788579866835,0.6304450829276416,0.04392551444280528,0.1153647953499684,0.4030880145760313,0.03567756947022106,3.151322628960288e-05,0.8005529239463015,0.001768608541175458,0.3943747556795172,0.01138641616213803,0.4249880408643882,0.1577353707778092,3.697117175902659e-05,0.5329931719427129,0.1280389394054104,0.8630368573284813,0.005265217655995512,0.8181068460520073,0.9585695664673159,0.006216425760012793,0.001429370295466334,0.3337738083292077,0.06815513156931161,9.012391956364467e-07,0.002706307536742272,0.001151710666159956,0.2401675051927137,0.2736064075248298,0.3685467416677955,0.09210912159736379,0.70558544842823,0.04295230065049436,0.2438366008161,0.0003560371355555865,0.05540050871643466,1.030968806665261e-05,0.006880474448511225,1.730858654669387e-05,0.3182438503943115,0.5554209379991504,0.5774732878916822,0.0001295006722720277,7.397360815817556e-06,0.9488293786984529,1.871296793743516e-22,0.004461706837559787,9.872538922019522e-06,1.595431483245792e-06,0.06807481765510048,0.04806541565370871,0.5780626100046447,0.0173114035389033,0.02203557700914266,0.9916170459338551,0.6128678725729048,0.6444873276633761,0.1016982214475762,5.081135487562797e-05,0.7712848002975813,0.4804939821676364,0.008800772739713292,0.0004177404994757411,0.002103129207788701,0.01329929019151114,4.521715442886486e-05,2.109663008648301e-05,0.2626737613994291,0.002017313392433339,0.001331955583654999,1.073717542449802e-07,0.8553262667766353,3.48105368084838e-11,0.002622419536263071,9.93535004705197e-06,1.49253031307919e-05,0.194750817341081,0.0003127508876906083,0.01740476882965247,0.4419521434325333,2.53140796300287e-06,0.4573518028684496,0.8181108713919248,0.01603943524147159,0.7538063510679722,0.6800125401359246,8.994627677060604e-07,0.3917744482489964,0.3294000598070869,0.0354736409104148,0.03485152891728075,0.8433509010467662,0.05796928138849274,0.697038916662684,0.01344978928811283,0.7310216420855912,0.01171748867167927,0.6955127508374442,0.01867958307090529,0.00974106479971466,0.8955006654104856,0.4056180750505992,0.001095496403373738,0.1587483439397586,0.5205252678194503,2.748334216108846e-07,0.706307428837764,0.01310210381253534,0.2302241721848713,0.00904952107786995,0.0277658090391292,0.7197024769794992,0.002446746942892272,2.176681425183635e-08,0.001932862957971725,0.6792605097426666,6.210945021295179e-11,0.7949196565666361,0.01484150199625443,0.7511763990839853,0.00125360531712138,0.8951601911895556,0.0007371044300163378,0.9192185515204666,0.6571992999212716,0.3113002272331163,0.05436055953456684,0.01124452343786207,9.67434043185341e-06,8.833631591713486e-06,0.8892665759905779,1.014829013900185e-06,0.01693937481824382,3.836143483484003e-07,0.001944087347645497,0.3614806943544503,0.3607155502893631,0.6947008257362401,0.02113558719364333,0.8242752653705815,0.05967213099560646,0.02329878773891782,1.397928945672832e-05,0.008664973121806956,7.194114774649118e-05,0.1002473659116109,0.009946221232918264,0.026697095953765,0.0994166548247807,0.1906805122840172,0.6649787284657631,0.4787128861190729,0.94403907717046,0.7487421736532451,0.8294954916541089,0.009723870017096398,0.001709985782291028,0.1175290593059779,0.09145640985439946,0.1074609706222605,0.005044916585679683,0.001880534959445557,0.06230455098335568,0.42522885249965,0.7524196837813943,0.5893331093589125,0.7631943391636598,0.01754017981509632,3.062956078833175e-05,0.2558418503082229,0.252101242460937,0.008176225790757148,0.4216179731223939,0.6624581024907739,0.6943269240078845,0.01574655220248182,0.03124910154857532,1.917192729894771e-05,0.5703162423176362,0.01310856898606191,0.006874923950784886,0.2828855604258803,0.05335903543215022,0.9452325628224987,1.397928945672832e-05,0.6104074124492969,0.006839072402094561,0.000744415649473846,0.004591501043457177,0.1068500034140608,0.09413364866003288,2.602820714482318e-06,0.0411011119700403,0.05427222342962008,0.3876918548587061,0.3738542255549279,0.01798873836244909,0.3867904718011927,0.2847556939430175,0.7756918544557035,0.1035624639438416,0.0004841534805512125,0.03090604176854352,0.00621206919681934,0.2666153251462566,0.3672475262888054,0.8277799026789704,0.02593216690718519,0.5971532939987155,0.2831244807679312,0.2237622133013341,0.0009256388746258095,0.4742622567827278,0.3370547390236703,0.8422800845997107,0.3950928788845527,0.004014574254198456,0.0001348910688780913,0.0002531092368737343,0.3397624141472227,0.0004968918532251506,0.2398864132623974,0.5263919592273507,0.6691492901535765,0.0225067382351514,0.01538995088455978,0.1382020530737617,0.954938950227045,0.09096724799754735,0.4882826093745577,0.4983716733325425,0.1379562693729147,0.6169686680500346,0.1368524203737582,6.199264473071528e-06,0.5037205149999638,0.7539575429989396,0.05275508299402507,6.11218237635565e-07,5.859631110658128e-13,0.0007547060153968312,9.74196930246934e-08,0.8907589925042412,0.2539394051212948,4.241815882978998e-05,2.056129224086085e-06,5.479717551240399e-05,6.432868834716077e-09,0.01611929313217651,0.7087266231513016,0.5646905918724132,0.004743550717796171,0.6623617198169471,0.03430869766990163,0.0001313649950374314,0.5482236195762433,1.196506182930401e-05,0.1295463102407595,0.1020247615207468,0.6161836087183407,0.6473057297781399,0.7575450194025697,0.02768270339164583,0.01599929481428937,0.003043765350250399,0.9677657230095952,8.507198340268218e-05,0.1188113249917134,2.511836572414616e-09,0.0005502905504314491,0.01978392828086457,0.5561074902788832,0.0006910794842488056,0.5283644472740511,0.0004202621804467705,0.1868574268258571,0.002244783831537209,0.01104100554953462,0.9943741956125359,0.3985963540531207,0.845482183912927,2.721919518097828e-06,0.006740020464543751,0.6531974319722681,0.5091199361905344,0.2191697892178552,0.1236090904340566,0.001064095832737197,0.8016178508002819,0.3397107892417183,0.2375413400522494,0.175856053543676,0.1723023905099811,0.5149685355752622,4.234885521062555e-06,0.4815097772956611,0.5009667981935684,0.02125852170118547,0.8542649232605477,0.6531362745209527,0.001134334684211873,0.06353139161766588,0.7271453855671474,0.0272661970409509,0.8658251649367662,0.5700831061325266,0.142302005781783,0.7542384685063431,0.8519509027606581,0.295197300685619,0.04668412096510965,0.0001765160465263163,0.7920231867865566,0.02787947108210607,0.2007007748565381,0.6944356402962001,0.3859101673971578,1.402170958244937e-05,0.1600654975989428,0.0002565216650194259,0.70558544842823,0.05733435533878898,0.1189616477089934,2.665025614717472e-17,0.9791358657966726,0.8500493579406452,0.1149569567514127,0.6420994281067234,0.1229876145599927,0.1006124617435647,0.009337668929488877,7.644534710655317e-05,0.7688865351287441,7.472812783196144e-05,0.0001679908253103121,0.5847548897367699,0.001188756786685886,0.1761523525887129,0.4960159443569412,0.0308264292752569,0.8940376292345515,0.002205133752995116,0.02435716659620767,0.8451735718849173,0.9912228914928193,0.9567280045814528,0.02896322605969104,0.877604482057851,0.2258730863254758,0.2302309868104189,0.3423111655401689,0.09697800921718412,0.002728779828425815,0.03967323370265666,0.9354126676843495,0.8446908723982686,0.0009628344123002702,0.1868574268258571,0.1666682258529429,0.9553308424446226,0.0009517238533526794,0.9787809280363078,0.288921632186217,0.02738910145884618,0.3563251924599958,0.9051138308967401,0.8332937900089283,0.0002147424166836941,0.1685195813332232,0.002816319006144605,0.01130101470978475,0.1002101794283396,0.02247791700296549,0.1821484249715883,0.8194115009614099,0.2310698805184046,0.1423810220095993,0.06690424062639191,0.003262692498047886,0.9862016960128989,0.7716378402425941,0.2608310918086705,0.004057909245011869,0.7662145280454294,0.3111957910769586,2.229053850818012e-09,0.09249757476310387,0.00436512509366073,0.5020616785353801,0.1234075088039182,0.3414789334965781,1.220367775849223e-05,0.7755859512971631,0.7965888150972729,0.9044254023895113,0.4493774369734016,0.9637842744281004,0.9926110422893731,0.9916711102768271,0.3213257458727924,0.8138725823788098,0.003572680610828767,0.2739270921305027,0.1254659122994257,0.01106483372968025,0.006532895923393169,0.005086819612042398,0.101383909029242,0.7109415994312174,0.4560957122830389,0.001034419638125852,2.253470944295807e-05,0.5592053118437813,0.020873012639602,1.270422540297586e-05,0.08158103150627598,0.01309639465968686,0.03493226697352287,0.3264513171104193,0.06430880156153239,0.01167242918432577,0.2170009984121338,0.2206856893780043,0.7273919557545322,0.2474932975299636,0.08908573316357292,0.5532885388710523,0.005743288446599111,0.05943633320232407,0.03530140598647769,0.001868133370855064,0.002644882030442253,0.01069684481915701,0.0127289177871424,0.2933884825668035,0.000711510921841876,1.001563395070084e-05,0.3376549673026983,0.170960784108358,0.6038908631915254,4.288369193904029e-05,0.1512983103702069,0.0003986502007670478,0.02919953920323698,0.589299017635095,0.8056993025566009,0.0005653416443301086,0.01091123389988809,3.555052132589709e-07,0.0001082988004069916,0.8333925178856707,0.7858526226803495,0.3624813870965586,1.249709270434686e-05,0.0601188641224778,0.02748304150917011,0.9666269165126723,0.002346138205214968,0.9524919743482566,0.1289617946741781,0.05758920360601318,0.3332533381388386,0.8682273278489838,0.9524919743482566,0.9205095066495995,0.7360360591809699,0.4619339991487293,0.02269059397983086,0.7186560201618627,0.2438366008161,0.03020440844794238,0.104106307496698,0.8288097648985422,0.001095496403373738,0.07962983920025581,0.8244241502169741,0.967171793221364,0.0002432230460388119,0.9087222719810929,0.007211848954183325,0.02409638003295942,0.06958318192353687,0.03561409416673297,1.840982345577752e-05,0.1009279029791215,0.4940523358932285,0.01097668290802355,0.766018254144878,0.7001931782632601,0.9053986336677005,0.4357644668171088,0.02706634754659237,0.7894776474325458,0.6531974319722681,0.01437612940584624,0.005980379895988071,6.186429369075781e-13,0.3806907730961141,1.869444640849602e-05,0.7668782774342219,2.107069064602799e-06,0.507001629710915,0.1331348281765018,0.04732503013423905,0.5162647127710539,0.001970958863156567,0.7779487974718282,0.4845758193367441,0.02052766513320856,0.3560311636350358,4.134869569379778e-05,0.007993530656745441,0.462692838052083,1.770812199619562e-07,0.002473683484392214,0.02141523309621859,0.0165720155180514,0.3778609051326162,0.6633171351243837,0.1590953880928035,6.491777909485e-05,0.08493108504141236,0.3193156187042698,0.2479419956205054,0.005415164556192533,0.2961378879587019,0.3554136998369515,0.9560674341093828,2.888221218886165e-06,0.363365318633732,0.940591757299756,0.00193955183205282,0.003368075509631545,0.08130727788542946,0.02204683001715399,0.003178987342028071,0.8208441737898783,0.3700492805495759,4.604572176629909e-06,3.329177553489546e-05,0.0001974586389518773,0.4213301608438767,0.6705655673559546,2.461841947514226e-05,0.252101242460937,0.048396140981978,0.1291592117974425,0.0003187257586732323,0.2626737613994291,3.149716676030612e-08,0.5060806026361515,0.3198915627323857,3.737936107894254e-05,0.01475651934177241,0.0001882899908316785,0.5733930515405783,0.2807523677619634,0.08704856498534347,0.01645372304828317,0.02622886120511592,0.2886657176630782,0.1813940037194557,0.3116069008182351,0.008318962996777066,0.01306730484842443,3.646592564773969e-05,1.795557314934412e-05,0.007019171292803958,6.569372377022468e-05,1.581286048792873e-05,0.3482244120224322,0.2603347186034653,0.3376273343482369,0.5728072647472132,0.5317424953674806,0.1122338696807303,0.0225090107798917,0.2207072372782128,0.712405920650702,0.0273010192752333,0.5418518977352316,0.07493557577609611,0.01136777895320348,0.0004748249010021497,0.9282757403679216,0.0004396507568202504,0.2186113592260311,0.292867405192586,0.0002105764455261702,0.8416070842721765,0.2079771135403158,3.30763987311842e-06,0.2406353100505265,0.005119145040819229,0.343813523795057,0.1906973389791824,3.2353782650671e-05,0.7772542237249117,0.3610208578382436,0.8739453652055261,5.547296342584586e-08,0.034789532257222,0.1749724459457123,0.1196561020319734,0.4038411845277134,0.001100708550274604,0.1552328253171083,0.0947535696281965,0.1854865333773227,0.9999364845311802,0.01432211170510217,0.09824180898375279,0.170550133881465,0.2066874418281648,1.441449887595595e-05,0.004057909245011869,0.7799346922041229,0.06154507843898502,0.001725534461036224,0.6054862830208223,6.160702486479616e-06,0.1821484249715883,0.05080738061428038,0.05758920360601318,0.001335250328797658,0.00260015862431067,0.8873527101384303,0.1454111778335714,0.00337540572966127,0.1164989817341266,0.0008864319292588097,0.46549255381534,0.001514720350107463,0.7797782609077046,4.025521046131898e-05,4.713507126114442e-08,0.4642895204890521,0.0001319293644971839,0.01166405098916082,0.1620981003624161,0.005510944133315474,0.9768490286946241,0.2640832848901888,0.7955549875846624,2.424080803785106e-05,0.7977287082729175,4.002163474763549e-08,0.1476257501865936,0.007172973532104683,0.1216699414909406,0.00254315762008296,0.4395477240900566,3.845610894725182e-05,0.8723868380994764,0.06942508243415384,0.1521819987281824,0.01091123389988809,0.02972599593211843,0.4643506191926593,0.07098384336055741,0.9303748110193421,0.02441412787200342,0.1125940988341855,0.4862829221924109,0.9637136924790304,0.8948616089190221,2.254518942486827e-05,0.7005711754717471,0.002011216358618641,0.2486446748876618,0.001499781526833245,0.07144882958658469,0.004178193270092469,0.5847007783291761,0.003277798591195551,0.5315800531774758,0.8149567827525669,0.174272958156751,0.321102650715461,0.6704628541423342,0.01524233022399987,0.05970649615235323,0.5516872626113684,5.027824574539285e-07,0.0001839893299828934,4.450837870994962e-06,0.1109605507496768,0.03378253602673272,0.001215586737728081,0.2884894789316464,0.9128336837614368,0.0002122836081110596,0.3978684514236178,0.9614736535031262,0.1567864638683652,0.9039735535263234,0.9901999461317573,0.005102013870357067,0.4535255712842046,0.406456107780404,2.177477058444724e-05,0.002149470769893615,0.7135835283019674,0.9680025018692554,0.08160628436633995,8.305302122857466e-06,0.2296466806671116,0.000741243541954593,0.02787890868281619,0.03079380503155032,0.1142561768309885,0.06651923676328332,0.1066123987707456,0.3040035419755655,0.2475690479841769,0.119959574371956,1.506363723125153e-05,0.475335879883388,0.6827722229792828,0.9915456355127263,0.5450665278085604,0.1926944003601921,0.005956039951035838,0.6325438841802982,0.2319659879292893,0.06017720407523958,0.007441854074788437,0.6101978612560164,0.007906134387300799,0.001558741211172566,0.02488110104411451,0.3425215148503385,0.02784074121210538,0.07929702170106022,0.4147733676630951,0.8935541565534381,1.279230644569895e-05,2.499405576462924e-05,0.1091595836245095,0.9192847104474219,0.3941386645521553,0.5807363502772309,0.4643506191926593,0.0768623633592584,6.399359245525459e-07,0.7214414435657972,0.02633144937115749,0.0001270290914355896,0.01627828209487422,0.237315171014994,0.5874014085805497,0.7056838401862913,0.08512768724742253,0.7182819055322586,0.5431832419984362,0.1381270771605892,0.9546679741895375,0.00375035093919363,0.2097322707153282,0.9060020163937786,0.4332739468320146,2.443356199972832e-05,3.307189989732111e-06,0.07586383921788424,2.491740085974205e-05,0.584393746289107,0.001126228660405008,0.0719790385413429,0.7472676672524994,0.2116785735305026,0.1485064729923449,0.0006202661422474707,0.6351943460337925,0.4211131211140328,0.8438001595498383,0.1810365010448722,0.05798774575905064,0.8190632143720433,0.1768300615298567,0.1308351860018771,0.2478456510851922,0.4205708413191743,0.01803487176002443,1.684631323727322e-05,0.001190466272301047,0.1454721823682632,0.6529374910576278,0.5165870938843206,4.185306115177702e-08,0.01509523748056032,0.8511593722080151,0.09591493175127125,0.0831841379566146,0.9290130706190274,0.1868524458704839,2.196068046170047e-07,0.004388851502922462,0.9340646862996124,0.9617441452006575,0.02170771150948655,0.1626865584745813,0.000641813598300011,0.1162267960414744,0.04017611176624174,0.004603198172319547,0.07015970849144959,0.1697335249510662,0.04907719422787515,0.002822044242561444,0.4787128861190729,0.01921569153963925,0.3861835479750037,0.838281589565302,3.27821899509992e-05,0.9585802618238179,0.7600726403364612,0.03319817138429936,0.4586158366789717,0.1251934572652002,0.527780183413476,0.360311443162397,0.5765058354451846,0.685339599623771,0.0002597648778609288,0.05103039014128079,0.2070838355819049,0.0006843188805964668,0.9129981546227747,9.115526614404055e-06,1.128186226506002e-09,0.05171168726172844,0.0268090129401478,0.8910399672511812,0.05763501455952498,0.1119143789691926,0.9271754834372264,0.1381015030246562,0.1035783279097514,0.2090469140548119,0.09063272592975193,0.3163501129762324,0.7101373987821097,0.07530388878228357,0.3105143249057863,8.960314380025785e-06,0.8248997958349494,0.09074426480147682,0.003280926502458727,0.3056264244251886,0.1003660084869968,0.004921590671417822,0.4382477891984101,0.8475762831286417,0.4243107465585528,0.002924875398324494,2.462363828837339e-09,7.063989624701254e-05,0.2010248598096623,4.319927597977915e-05,0.1485307440179709,0.004421680429829032,0.1097575162918341,0.06029666904297419,0.0008052827260482719,0.1588549503819866,0.3197675688978915,0.7120325970986099,9.441351561424608e-07,0.01764032584226274,0.9686063338822,1.100754077318754e-06,0.0001165277980800012,0.1133105596002472,4.501661270669388e-07,0.03967323370265666,0.300951553352892,0.0404768795675808,0.112730776355465,0.7236256603141121,0.328681669938007,0.0011708684130781,0.0003162241020936728,0.07754931586842544,0.002351146348208757,0.1841716345691143,0.957214834460033,6.035248428551369e-05,0.4382354497936734,0.9399978837138154,0.3596300192620709,0.4249955328373319,0.0004663325338163414,5.734387111625128e-08,0.01475427558423396,0.7689714695996296,0.008303659926975538,0.9219679507788655,0.0406212270691542,0.8437344137030092,0.4874517902836781,0.7158194992430056,0.9835405744410527,4.144263276856218e-08,0.7894776474325458,0.04753259149322114,1.303258418509293e-05,0.001101135344062372,0.791769762138153,0.6497213725458773,0.6926157488206697,8.270572175683196e-05,0.002087045603794464,0.07591823910910336,0.3816681933378429,0.05757608816192736,0.1096152190627406,0.904232470463803,0.01502348542132171,0.2302309868104189,0.2304317319748622,0.5869193208712619,0.04375157597661909,0.8466977292386075,0.003830808386874395,0.2158180408004891,0.001142577017716024,0.6906568482841626,0.007460563930861008,0.1488230020056988,3.333342706283794e-06,0.6562190947335926,6.92520453502302e-05,0.9334972636748305,0.0002072722368713514,0.4175672530064892,0.5462261656190424,5.080142567687927e-05,0.1664958965104908,0.005443162399429264,0.2566013235917359,0.0002526346041334291,2.557194987851437e-05,0.4726814729460603,0.01301463431483142,0.7700483900328142,0.4792964731496627,0.9527117666466172,7.484730499756385e-10,0.02208426070926452,0.0002555245819720389,0.6556158867943204,0.4222985603152958,0.2591055893144781,0.2728758979571717,0.9465753333513214,0.7745327958319056,0.8680241263615879,0.392801886106639,0.5279409481245841,0.04818195215962696,0.535787787882539,0.01063808226538619,0.7289706590424464,0.5018730874998326,0.5162647127710539,0.7272998603716332,0.04657258365155263,0.08095158986136332,0.4411953682597536,0.006628473381915201,0.08533575891055954,0.2769622755684615,0.4337778198873903,0.1564414096051256,0.7401274848342255,4.799277214680917e-05,0.408199859109631,0.9510177664294567,0.4224870881718565,0.7641975331731772,0.01186194949569247,0.3581112192629415,1.522314064559977e-14,0.8131252067577595,0.0004366460668623382,0.004687626988759054,0.01171748867167927,0.303674269675426,0.7901680891640855,8.737028773156475e-07,0.8188621258397365,0.7452533852104876,0.0114903230503044,1.278641955215216e-07,0.0001408939066058308,0.981795622538029,0.1460902607630815,0.3950357303756067,0.5423738124011851,2.228332897330293e-05,0.00492762134518897,0.0053483021744998,0.4807389358689165,0.1445662290847234,0.03127570244539716,0.02560014550690385,0.2535867284098091,0.53205375180383,0.2009338607468115,0.008043965650617123,0.8037938386243559,0.3360544752893428,0.9969741606075351,0.2843294550432842,0.2602644481476083,0.02770831292613455,0.08029029137440856,0.07068805796690851,0.05380063006718164,0.8295810113300842,0.006078175901067,0.5700831061325266,0.9809637145154572,2.622524902925918e-06,0.9638948470588448,0.821826347808002,0.313967343188005,0.01768648864392848,7.771884126688867e-05,0.01508164363309763,0.03330629023284533,6.969812241490599e-05,0.3465758290532857,0.8609507870379646,0.9885810235924843,0.5909571021914438,0.04482153703906253,0.03091420678328313,0.004899479228075113,0.00605739234120362,3.697117175902659e-05,0.05057373456912043,0.03282605831902333,0.0947678465643946,0.04028507459842676,4.090878472935575e-09,0.003107797113722167,0.06552916721308356,0.002589721063152977,0.5060806026361515,0.05098494921425314,0.0001221979473966235,0.1891530990547475,0.006756661681416516,1.105693241135156e-05,4.232179994175347e-05,0.02052492603383244,0.07775280659276744,0.4209136613998921,0.9722196428491905,0.661995536050469,0.2815843134459867,0.9603014331411085,0.6136192797782061,0.05171168726172844,5.186356148075975e-05,0.0560642534930878,0.4983716733325425,0.01443500248181925,0.01245679173360987,0.1122156344929332,0.01242921600267531,0.8415883291920327,0.1536196968943813,0.9098278533952394,0.7590674946752155,0.2188695135251888,6.890506619961819e-05,0.01573405363841182,0.008237222758542266,0.9879005831809026,0.8446908723982686,0.9928813072194947,0.1512682193317474,0.4600044660689976,0.6264106449397931,0.8811348616346973,0.3190837512837917,0.01847020444922704,0.01529620072974055,3.049590350230806e-05,0.2500510674211911,0.13612055009724,0.2590353445481645,0.6408172828474565,0.2100082956773172,0.8395577584309113,0.003791547124629974,0.03050025618052642,0.5793737155578007,1.313519871247031e-05,0.3365034527003161,0.2746062141368557,0.06771487844326395,0.8704317111022358,9.968937938512719e-06,0.3008344825475054,0.3280583450506659,0.05790874738004365,0.7504698150773303,0.04996899811829311,8.154985898295828e-08,0.7432588393072754,0.003354479668780276,0.01295920315761208,0.4947692212166012,0.08498258832936786,0.5060806026361515,0.9663631288598223,0.4370507161608043,0.01827508253871871,0.009244530256273968,0.01528835197137105,0.8898768534148275,0.09577235140151108,0.004809025649911238,3.625618985982334e-08,0.6826138061698256,0.08226038549804084,0.6541714925655027,0.891131492131317,0.7159522863796922,0.06313393018911935,0.08461354694405582,0.632796948574615,0.0126216740474152,0.06009803582699273,0.4699166286404944,0.001039695861665284,0.2539394051212948,0.6867777556276579,0.2214716898479507,0.1267240671662807,0.009325869519132226,0.04003071081508572,0.08472419517719287,0.006073045322341993,0.5203963149818421,0.3856827079785159,0.2087263630586912,0.122896268388894,0.01582097318210806,0.007458403615598771,1.99056429856091e-05,0.232325613835414,0.1723435577284638,0.2592259332430168,0.0857613404817867,0.6827722229792828,0.8749157495311396,0.8240057717990602,0.0006752636753738956,0.1797697610722665,0.9834493731662909,0.004210358514241063,0.0003116516435772564,0.9108824286521974,0.3671327717306888,0.2474932975299636,0.882676011695239,0.2886534239545057,0.3095861997096024,0.001075362620224226,0.6141965272956401,0.7253641196711846,0.04332308000266512,0.06017720407523958,0.3440542314367259,0.2567708877446969,0.8919277655371299,0.08461296094161561,0.05951505012945902,0.6072183973567098,0.03822749291262847,0.02249411936816138,0.2615140518338034,0.01000739683986494,0.8281602773301449,0.01207069159529324,0.003894259859726141,0.6171371141414943,0.05156206041248487,0.2666153251462566,0.1296523270808501,0.6283544948830397,0.01717127117674549,0.0003416638164282657,0.07304052817791673,0.002917673977941619,0.193192181653711,0.4846805755579523,0.8372375508605988,0.2044507659533973,0.4582662143521904,0.1082165593670901,0.644365526930151,0.1131765430709138,0.1695494732021268,0.0002289138658773586,0.6914400321026339,4.350380484359267e-05,0.03098592282207885,0.8179324677617908,0.05682823175391329,0.8623279674327174,0.2232168773360037,0.0001544723637306358,0.6253876628777095,0.2624883216560871,1.361173665174831e-05,0.3442772274798881,0.01873674459052944,0.08613360589124897,0.3376273343482369,0.0001189644355859959,0.2820292136117329,0.3107251261707492,0.6497213725458773,1.073717542449802e-07,0.5206060294065511,0.08955579541609134,0.0004971830559092858,0.0003597209592093572,7.671063185364306e-05,0.8086323513306397,0.8539321721329582,0.3873506673230663,5.535545665826859e-21,0.3294198849583681,0.5122540737181379,0.4402631540360702,0.04677375461258278,0.05582194545388812,0.8460916042616718,0.009722916813833736,0.5055000186372901,0.2826196483999132,0.002393562104207949,0.003289135246722361,0.00173034657668822,0.04807039070158093,0.1567864638683652,0.06109372168883557,0.4279880573433598,0.1128757740587071,0.9423189960178995,0.7551003703464393,0.3003808398448579,0.03033664347076793,2.188415633539761e-05,0.1944762134458819,0.03977992365525656,0.9572036065632887,0.6779049889984184,0.01705473499459906,0.6986542641793809,0.4888095759206237,0.0002172168179496224,0.00786427646703306,0.6531362745209527,0.6091918273905667,0.8214358870892194,0.06619493602092411,0.01344300149631444,0.3206070390201082,0.376730474080302,0.1651783773301631,0.338656482729815,0.4636203792859804,0.007229043296294725,0.6543623219950002,0.8689828680530113,0.5638894936106927,0.5662334264425245,0.3439875690819564,6.11119540244433e-05,4.533886134359315e-06,1.237151517460578e-05,0.003319822906566957,0.06428211804322413,0.02299493849493858,0.04341943610594633,0.00118536091482842,0.1331576583784015,0.5978462880007961,0.3310376868834192,0.09274559460724627,0.1235285548968118,5.028919190012373e-14,0.891131492131317,0.753022655386179,0.02629221563524606,9.427081893081373e-06,0.7717097827887253,0.008711824046959575,0.8054115326014261,2.104480417804951e-07,0.01171748867167927,0.1225633372725347,0.0831841379566146,0.8138725823788098,0.3990625625704046,0.6809094524139143,0.76656905581375,0.9068863833064683,0.8976903128919661,0.6382332607027866,0.3201407808318338,0.0206360039075175,0.7715267148079097,0.8992379742616347,0.8833948363729157,0.02956721067092518,0.7271815456095324,0.0360728216886597,0.0288159950718627,0.2549211165088527,0.3653444084063794,0.8568862452067008,0.5693563858202286,0.7815576867706236,0.004805997411323164,1.40353414055407e-09,0.05719921469917617,0.8827622376909376,0.05471094853704851,0.2016591174234154,0.477771334859289,0.004569563521671282,0.1082319816979846,0.02878588142437173,3.477213339332275e-08,0.01966661431741193,0.6353072062146693,0.02897632438381035,0.09326931546747171,0.3650529857395265,9.340514126845577e-06,0.0823315828390425,0.001124555958539504,0.7831318944500014,0.03488626534638974,0.3904642907452636,1.346792051168899e-09,0.006181362963240661,0.01999184707815077,0.006487593724726159,0.004408359734534715,0.02006992048038733,0.4742714768900432,1.80515477722218e-08,5.843493145520748e-06,0.08106956786908597,0.09913037331005423,0.9667563238836324,0.002110905930519638,0.2067048897072985,0.1088655460931745,0.6677737513523793,0.8793622296611284,0.2425094021647648,0.01198996775806779,0.9199901973844509,0.597180997490325,0.2824722032864976,0.1632211385291114,0.4749266495506336,0.06154672772647077,0.319445152345717,0.5605759507044032,0.005039650348331072,0.09626683529850336,0.08770757050616447,0.08942340829401499,0.006691841757057485,7.800706922701108e-12,5.44717670391561e-05,0.4298245249287646,0.2176339964825929,0.08090414918261737,9.44822022105287e-09,0.6699408684527994,0.000933464206684146,0.4395477240900566,0.9024924365652234,0.1506281066799662,0.6747157217461009,0.004871595616430557,0.01759648244445261,0.2296466806671116,0.9975985768785923,0.07216669826751368,9.998647403253479e-20,0.1119922721504805,3.939259330507901e-06,0.1785711633599749,0.09391527682282685,0.167965284853924,0.1105521884681247,0.04129400198312628,0.1029215290567709,0.09249757476310387,0.4008849540289656,0.644462663852934,0.09330606292358848,0.426978514767022,0.007172656589152887,0.7166630783696201,4.541403214362366e-05,0.0001723366927226554,0.0003711129156308601,0.2603347186034653,0.03612075392514214,0.736294258669271,0.5417844250867309,0.07245251679727613,0.012136861839293,1.706995080516106e-05,0.5628834384418213,0.007072754462991162,3.306430373842624e-06,0.1879498365130004,0.007471946004185164,3.329931672234032e-12,0.03947617691837203,0.5359707954413061,0.9882496511809791,0.2958238119667142,0.6199599932367441,0.472267809625374,0.2650746701112559,0.1410731246482471,0.030015466791938,0.645792138833635,0.6988817987738446,0.8833948363729157,4.76919804386765e-07,0.7534663889935825,0.5121003998523723,0.9157883830813817,0.4411307403649212,0.9233161040252401,0.02089800114863025,0.0206541451020181,0.3197867835433836,2.329360202434137e-05,0.2301241469857813,0.001675629381076889,1.233705593598595e-22,0.3018483975635461,0.1186434283787427,4.740906373601423e-25,0.1943556776471635,0.05472936724759334,0.2839976107122642,0.2221865583359649,0.0002489523696305838,0.007466241032653881,0.1191993692321543,0.7878483052400369,0.4789023727388315,0.001416626276562649,0.007372076021642263,0.4989258230188183,0.6045017014769808,0.05627533481923713,0.05810059186152192,0.5894951975520228,0.04280569629810523,0.9140495083200286,5.839048728415975e-05,1.895572957663047e-06,6.506660173996748e-07,0.001130323381210934,0.903486632302863,0.1636050318127314,0.8466977292386075,0.8584149865236252,4.692520670389858e-06,0.2818716577729954,0.009577519728309239,0.003945153972783852,0.003750700770603787,0.3950000842124223,0.5896926863515476,0.74122660971058,0.006143946248974653,2.307861316290088e-05,0.3119503967993608,0.0001142688790036455,0.06754197261553031,0.004261654319136276,0.02834352881048322,0.02086832618696924,0.000188223212801374,0.4176876223258247,0.002748984869009922,0.0003718667918918406,0.2871283990977446,0.0009972339985700454,0.4727328083309673,0.005168889496962829,0.5507635586401322,0.2239198791011858,6.556826590634355e-05,0.2395648003504091,0.5139746536968679,0.9233907489890681,0.5787162048536032,0.1409070919435667,0.001952575677561559,0.2613817097313676,0.03857031965286204,0.5689168538620649,0.007628166605540184,0.7963221101572802,0.0002006421343613348,0.1724393107402013,0.08042515974539252,0.008506547078688795,0.5508175163401222,0.08710449519531127,0.2877068581717097,0.9167416871882991,0.0221632168657353,0.6027138589901989,0.5910482531893145,0.1006368131122609,0.2094285857885442,0.5930744002589982,0.1049944222646817,0.1485291077260198,0.008927473691629271,0.8712761573271902,0.0003795944960078059,0.02787890868281619,0.9255467956312274,0.01431081489813677,0.0002182433586854922,0.0006667450032623984,0.03195704109872479,0.9416143797356116,0.2869960541453584,0.009535940110596395,0.2912032337548613,0.5282958689185843,0.6820083715041171,0.01104100554953462,0.01330864895995391,0.3941236567628255,0.8422800845997107,0.105004435965787,0.2475105047022181,0.2622605323717488,0.5694585528840701,0.7589125152605171,0.0182534965751593,0.001204779588036796,0.02116511924161986,0.9335429134661165,0.05456580827433093,0.3919811861738008,0.002971621579432915,0.9873577815023838,0.5962165692651199,0.4841569730598212,0.8951356288795886,0.4495935852005704,0.00781383680453065,8.616218448023653e-05,0.05455101498713387,0.4735534724656382,0.8415730103256502,0.5223490583525926,0.3873506673230663,0.01532443849377301,0.0002589237586987312,0.09107933823561803,0.00580069883985365,0.1427408831848306,0.7689714695996296,0.02083281668456728,0.003537077401295808,0.9615856907698053,0.1229876145599927,0.7890609655853004,0.06864984649117105,0.1485064729923449,0.6857262921358421,0.02754771662111112,0.7194545149350857,0.6590773652780622,0.0009618478960172522,0.05463483051880431,0.4595696897001346,0.2314224343239757,0.1572920670201685,0.2655766413698205,0.002164889468558755,0.2558418503082229,0.01591249566665812,0.3385325459669716,0.0001091276996464839,0.1048420261583337,0.9188370457542147,0.003027831736349833,0.1398200660014431,0.6923506721253067,1.666019915502879e-05,0.2723225404574259,0.07948583397017117,3.88363737581439e-06,0.03660373215174267,0.2391357604689532,0.8323216869098786,0.6837548337187409,0.007823434608484222,0.3741712948046189,0.846766036919922,0.5746401193926497,0.5871941198699995,0.8681341977914266,0.2652370333009643,0.0003833828640564619,0.03208609277983776,0.2796758657099834,0.6959897188675029,0.001512606295496837,0.713220255235972,0.7871448737116492,0.001051929884455066,0.1410731246482471,0.9686618370976594,1.251847273728666e-07,0.6290888415512228,0.5570171064822925,0.08659059589843288,0.9667249776203444,0.8655743720826593,0.09249757476310387,0.5853957555616968,0.009655795566902162,0.0009821114292338667,0.4716189406116641,0.4098598342599275,0.2434253515790368,0.05889674645378782,0.1106476640627402,0.193084642842388,0.9826357374364547,6.74849474219571e-06,0.1898488635271459,0.08503344950221095,0.5835911734301246,0.1008321626083957,8.621225980639935e-09,0.007597246163311396,0.0003711129156308601,0.8986701871793643,1.099208797530951e-09,0.132037444356954,0.7448619276795115,0.4420344946469559,0.2941709282165308,0.8937085114361101,2.379745949275126e-10,0.009910847434804203,0.7073855377038589,0.0002946040590136737,0.9817501218093501,6.204651371498439e-23,0.005022865302128996,0.5263177521536224,0.8243829089444507,0.9732694137993985,0.0003172724255406536,0.2256356542793164,0.2214537179994338,0.06972353987566381,0.001550381515742648,0.9376431873634741,0.0009012880840947016,0.3385376516913801,0.005767468424185686,0.8236063512906088,0.007801585841534857,0.9991963623477245,0.05220899541337683,0.7385119317886562,0.6564003019227062,0.04269841524201711,0.1113341578934525,0.98834910872671,0.3911247096821564,1.82845435117913e-05,0.1501069675222724,0.05485249451945996,0.0002718085204641408,0.9344598995776227,0.5005625542302686,0.4676946208322098,0.6788264507371096,0.6914845800927206,0.03665319619058243,0.009232662463910262,0.0002421035967547778,0.2397710683542295,0.1724428919173225,0.04341802301559013,0.000711510921841876,0.7688848764026552,0.05194868686598756,0.480980784885318,0.5382986258140205,0.01048436009293694,0.1399741834231116,0.1331348281765018,0.01026649829431191,0.8184489812899766,0.1748150936409668,0.06196825527811164,0.3344089354955957,0.7209306749072952,0.4947075002767427,0.4482137150858864,0.7710519951276995,0.6320093643074581,0.006538829867732959,0.02449523114892803,0.1482149851014954,0.000690858621588631,0.3563307222009632,0.001110762740791741,0.06386911116168523,0.008943332458402476,0.2395648003504091,0.7623137220936745,0.01413274939852924,0.0837924095084519,0.5380251141310698,0.06992183925829623,0.1191671856861514,0.5574149501770199,0.8138725823788098,0.5612784013783708,0.2566366774748292,0.3049212874160611,0.0266105135751496,0.01886600475430071,0.0042991179749628,0.0183186117411199,0.0719790385413429,1.218122998520199e-10,0.05987265975587301,0.07312243612168461,0.1636050318127314,0.2556458472495933,0.03857031965286204,0.9197334197918003,0.9488068977974551,0.4403399767047642,0.06428211804322413,0.7106338171509219,0.03754440834812497,0.004959180660201354,0.004125312784870714,0.8898142649496492,0.9787087453617848,0.9488293786984529,0.6422569331937285,0.02039424152528954,0.09619062696501177,0.02316711633774282,8.945854337623198e-07,0.0009371974490298744,0.1435012008069153,0.131058460247219,0.2857436014645142,0.1231585416718836,0.05360249000094939,0.169102932551719,0.0001840260799371224,0.09019409688346125,0.5163793287046943,0.9741168099113968,0.4304081784334264,0.07792333701335759,0.01297800408557377,0.03385576117957812,1.506363723125153e-05,0.5002899115854653,0.4477698367632784,0.1869096362727519,0.1044552309266806,0.009633788831384353,0.3890740927628228,0.02598734602754169,0.4731851772856817,0.0002955435366051787,0.6595503058984481,0.1048420261583337,0.1277735791676522,0.8833948363729157,3.724010614673807e-05,0.9362235521451817,0.209616384902628,0.01702128625729768,0.8260024114821645,0.6215877094541781,0.002644882030442253,0.7378739690915429,0.002685247238637407,0.3309533116538398,0.001168304961997119,0.002192934390604757,0.263719029042171,0.2632344958028394,0.8900179546033162,0.7096168090622836,0.9581317643144702,0.2313314338215372,0.2977854617880292,0.2626737613994291,0.2446816499511603,8.374300048806112e-10,0.4372098581403747,0.5720009956490045,0.8228408887318165,0.6649787284657631,0.6015467811491439,3.836102128530594e-12,6.199264473071528e-06,0.1932475747378198,0.001896962242657549,4.746219588611878e-10,0.07595428699324076,0.3617327789584291,0.2076380332871269,0.002264728700486181,0.009535940110596395,0.3423111655401689,0.0002505736869913943,0.1511878599127607,0.3014916617596965,0.4440765012619233,0.853565409645084,0.8553155350262034,0.1218370173625186,3.623141675811064e-08,0.2131166195626422,0.02069091442079157,0.8952600338464264,0.8843249868668023,0.07786313554216563,0.9567280045814528,0.1093575959542118,0.03004945443465066,0.01946116478927465,0.578818147945045,0.72870995840994,0.2245087120548998,0.1496941821401064,0.03737188103818982,0.0002328551746450122,0.7664827833276338,2.922054775997834e-05,0.6029069018303872,0.2911573476949791,0.184797254808849,0.9953662136556667,0.7149423006205731,0.4463277546969384,0.06004494455168718,0.872840099983349,0.001486340621823306,0.882676011695239,0.003461978682128124,0.8628639288817024,0.2775165801323667,0.1512745071108363,0.0001270290914355896,0.4593410357649612,0.2935112583956757,0.04418249325099648,0.04515469688140227,0.8930044589746331,0.000271406531714454,0.0001735050692561855,0.3048080861920024,0.465597477607034,0.04096311950002997,6.077745817112545e-08,0.818747165863571,0.4634765769025929,0.0139148618508446,0.3465758290532857,0.4673276385558409,0.3739889372191548,0.1494792908706646,0.3495257841000829,0.02913840516282491,0.5037205149999638,0.1999986655474812,0.1345875638053987,0.4979393606029883,0.2197108703024903,0.7644592860765984,0.009452298957613469,0.1798518414372118,0.06976869261185098,0.000159966885095445,0.4138312896559256,0.965386520611962,0.6562002579987023,0.099674432851481,0.1168957857479524,0.6734311978001321,0.3131487093566729,0.9192185515204666,0.02051370191937414,0.4732452923890678,0.6085250366453875,0.1085874134950669,0.02661250923918238,0.009383780269105449,0.9499058844169334,0.9834493731662909,0.2817713156910994,0.5313613700596647,0.05650642397994675,0.2776957824425338,0.0002727214269884317,0.2102328632718577,0.9934425175554431,8.021089946030685e-07,0.006395071964426731,0.02273117285354411,0.9735491424060427,0.8489651695356301,0.001274107024574402,0.005674064975935571,0.05486966327629648,0.7441391648846232,0.1569797400861699,2.815143559205831e-14,0.543872487343095,0.1199485042568023,0.7715267148079097,9.32551315610392e-08,0.04604499700694239,0.8031677058165605,0.1128757740587071,0.569528801545102,0.02537367610500846,0.005532747517875137,0.3286400037417262,0.5932604075262662,0.00219867794597317,0.9528871196109612,0.1229876145599927,0.08288667270969464,0.005282041429837308,0.2050324824295983,3.4309435485277e-06,2.304572954950439e-07,0.0003574938901622628,0.3277496035721955,0.04803972306852134,0.4098699373760809,0.08641863027381094,0.685477989836521,0.1041616005518773,0.001602821839972163,0.05045708902788224,0.8469562149725246,0.3419828246183593,0.3738623837895894,0.4148130078315604,9.729474656317405e-06,0.7823541735050383,0.3472210047759364,0.07025436829969336,5.477401948519854e-05,6.760884640667794e-07,0.000191396851729915,0.1502725674147877,0.008316460197520413,0.9213184440884313,0.1503611502819016,0.09882940657491146,0.02661999532460104,8.54193707723513e-05,0.0008674451139591766,4.680169106989102e-06,0.06084961335240686,1.572301497792217e-06,0.9316470433649914,1.249709270434686e-05,0.2095805038985048,0.2541756547047221,0.02094286447036438,0.1095594723302204,0.9238851059101465,0.2172723795338385,0.0006901374753241941,0.9431118397171249,0.3856827079785159,0.9319033734380527,0.8568862452067008,0.2032780799048832,0.3576903484961255,3.909269107336161e-05,0.1658434326363331,0.09174374115621382,0.006511438459063402,0.9534553094741761,0.000767566045898234,0.007026398353698086,0.0005762769076379572,0.359412214980678,0.6325438841802982,0.002582665830559923,0.001218782933205942,0.4039477666932791,0.02864330027884276,4.844928066054742e-05,0.001524116776893743,8.633444217665884e-05,0.00943469509816809,1.643591480563512e-05,0.374111187819162,0.4424075872020474,1.388329171541043e-05,0.3122343072767619,0.891562178225777,0.2270018193616079,0.3608104380149014,5.555969552520366e-05,0.9931593647709804,0.1360629914994124,0.14211539560584,0.0002296514826606513,0.1965968328299861,0.04214394170947414,0.004823560241212193,0.662508143599684,0.00381063760885226,0.8565676596674524,5.367338783549918e-06,0.2397988817140284,0.1587997539758262,0.2916351944936496,0.263569416583983,0.01291481227212674,0.420149540742596,0.4375867726236236,1.881519494563833e-05,0.38644517542041,0.7001931782632601,0.01154276481002982,0.2827530832556391,0.3884673228481954,0.1096358901062499,8.221246838872732e-05,0.4403971911021426,0.8016178508002819,0.4873049990213263,0.003942047750118449,1.80597478245666e-05,0.219394633801761,0.3809648921274866,0.5630711639349798,5.080142567687927e-05,0.4506703524668473,0.9667249776203444,0.3006845354166801,0.7436733556548518,1.073717542449802e-07,0.08252385629283858,2.180029950575453e-07,0.001436544956467208,3.151693420659563e-05,0.3080793735661395,0.005659408294586657,0.0744227538527307,2.739834716341492e-06,0.5595353595381842,0.0002737034694692983,0.7908527548093673,0.2362372056812083,1.337474377762494e-05,0.01181587130005619,0.2048345337496913,0.02142091013174422,0.3577431199617733,0.2202518991600624,0.255777146398861,0.1412975134957791,0.1193259841743207,0.0007333441603836878,0.8832375185852381,0.8249243555326579,0.5250742088248416,1.592157776882086e-05,0.09609837053435831,0.6561584022645123,0.9934065214236398,4.997592559721215e-24,0.07140069949907268,0.3135879373685328,0.03746675888428617,0.004042980442234153,0.4220067851084847,0.7819687051621176,0.02354117814108236,0.001479295663099599,5.50524532984167e-06,0.00332933622094815,0.4861170709622915,0.00645351231193663,4.648237467901375e-06,0.0009569666850380236,0.06526988996857373,0.02198229041884691,0.07304052817791673,0.003620794714168626,0.9963043789093764,0.001691938422113464,0.231204077094934,0.6940732534165956,0.279390561007644,0.4271229537642844,4.258942891102566e-05,0.0007866849834215475,0.1339004620217886,0.2088146728151614,0.1380590874748095,0.000327220105234956,0.001875906968302579,0.0001395381535596,0.8593868186070414,0.00297217437193776,0.5144629114972372,0.1454721823682632,0.0006076053889310919,0.00113918812166868,0.0002850100452076543,0.4345573604486307,0.0006297787551108641,0.2571431895832408,0.06912362795252523,0.8428627845903675,0.0003238396429835498,0.4188713275447348,0.01199852880331802,0.00482463254157375,0.0004569980171482811,0.00944055534548025,0.01413065183818941,0.6970120457429227,0.09893739458381987,5.003292995014943e-07,0.01522509074622407,0.01930725691912104,0.4899836701749448,0.2279477988057523,0.003211472118821046,0.373876219735863,0.1235088426904601,0.4123639719407735,0.7847917322263428,0.2007879179679022,0.5835251536776148,0.831979904583714,0.9475731803397907,0.0004873665273611155,0.01860191248901135,0.01828866199673575,0.8546144860990414,9.281076150124052e-07,0.01499383336619971,7.032306524479539e-05,0.07880358000376375,0.6906543089424633,0.8174652706922432,0.2281696009769879,0.001532483092399595,0.4242863564631523,0.6455227433266946,0.001652581365004268,0.1859556332457228,0.2511693792858149,0.3413443457625713,0.6996379389755902,0.5857160168377828,0.2797789290478393,0.775047555327687,7.58351026668209e-06,0.006327963927838568,0.001139018074119801,0.0001782083898401347,0.004414253929033093,0.06351189288500042,0.1221027921719175,0.1705892729126216,1.504924679095393e-08,8.552944887769649e-07,0.5820841850719317,0.634327347188973,0.6107307994717246,0.0131162067618627,0.01076217349688606,3.167033597563595e-17,0.92273907670411,0.8260024114821645,0.0002131786605031836,0.05357233377589301,0.03428753829656427,0.04909196993762387,0.3602017111023277,0.6178162313986855,0.1655674888971508,0.4035006722604049,0.6088004503264968,0.2536755400053454,0.1460902607630815,0.04615355004191584,0.04330591398408025,0.7805666358072886,0.7927953408385569,0.7209511970999812,0.2923461779365777,0.0008032499682948076,0.6318992226973903,6.503512238680688e-08,0.8804948997870423,0.05998252334580743,0.5019063689928843,0.003816114981624132,0.03501038707978337,0.2884894789316464,0.4656513939840234,0.8759581751942805,0.07705037426573697,0.001064095832737197,0.6891316575068417,0.5711179940222767,0.3139794004206753,0.1610861128031549,0.8655743720826593,0.4016057074914142,0.6732918035308934,0.6978102311787022,0.1184898960339169,0.4510635763272967,5.014539180903782e-06,0.002881265476422423,0.3546156493198778,0.1607349181556073,4.000115735611104e-07,0.5287267800149633,0.7398629758541602,0.1008321626083957,0.5730619427463086,0.1938208348073516,0.02316711633774282,0.04949377980340548,0.3162571512999724,0.08781567758095631,0.03379583249548217,0.6788351428133287,0.2369831964884495,0.092789558859902,0.7111107147292368,0.01803101035945011,0.003048475033847762,0.01200058973500383,0.005295221707278017,0.8468880999864162,0.03358393980208019,5.186356148075975e-05,0.4941869278718923,0.9316470433649914,0.9099752110649625,0.5418518977352316,0.279390561007644,0.06004494455168718,0.001445212720127164,0.0776717588375482,0.1649244489379334,0.6593971905764445,0.0003406042616908562,0.04203042804489142,0.9902905643636239,0.8982176716853583,0.0005570531939074446,0.0001926912769690579,0.3189692221911953,0.9837098976645,0.5294569344202658,0.797336529035946,0.9995246520617577,0.1278468804444092,0.02124780368508504,0.0773469930308555,0.9079122179237392,0.1083207258892477,0.05967213099560646,0.00539279043258441,4.63789121456145e-09,0.9561410619390153,0.7767756444746721,0.03977992365525656,0.0003781589035731059,0.196377786432497,0.05202115966333867,0.8907589925042412,0.01173870748386113,0.5683046345843966,0.117522476264081,0.6488621302452563,0.2331394841111349,0.07124491830273613,0.8149751051691113,0.01525037375160877,0.005422392176027848,0.2176178805156357,0.02239865319818957,0.6871034976264206,0.03834310451491126,0.003454796052554979,0.05416064965676717,0.55313648042341,0.7544422931545985,0.4877801835211611,0.184797254808849,0.005793696342452723,0.321747707901364,0.4008202570719432,7.472812783196144e-05,0.04395109345025938,0.228898662944458,0.007234802113255978,0.001565778596952048,0.8902321277651541,0.9672819170575541,0.01322461124380143,0.3368406220889942,0.6167427945919683,0.4368656646261094,8.687775195176471e-08,0.9015245125758632,0.05745002150432397,0.4656513939840234,0.2112700165735877,0.07775280659276744,0.9350595769451101,3.939259330507901e-06,0.1777188699448592,1.089581937623946e-05,0.000623562426360559,0.6800523027740394,0.8736766239628974,0.09767359903005736,0.6987942635554578,0.299577857791561,0.5626635307428823,0.0249138900245714,0.02661906971490477,0.249504056560023,0.1242588667333162,0.8429640734532976,0.07079654831763545,9.863077014566551e-09,0.002128236954588257,0.005032086837036621,0.1258424753869613,0.2005882839459069,0.7085051483305576,0.2406353100505265,0.0007118722571888402,0.5479636773913158,0.0340195020127301,0.1687494598097959,0.0006724471778221353,0.5291218627774736,0.00052031596641664,0.1523868577947532,0.09254635138857564,0.5967277656411298,0.5246023052305091,0.0009396968046900848,0.2565260287285688,0.1113614189137009,0.1353416492775449,0.01585100917439998,0.9964906732910624,0.0225067382351514,0.6926157488206697,0.8397818612856659,0.07721440671729286,0.2598196610733036,0.0004815304671962731,0.1954310892736873,0.0003597209592093572,0.2434253515790368,0.03338632564655705,0.7166630783696201,0.1171910324736454,0.3611067928513189,0.4039477666932791,0.002833068046457967,0.1149720055481498,0.7086789148157602,0.03221967143435773,0.002185187401477703,0.3759895061236151,0.00644365915042467,0.3876929978116246,0.0001457689790437158,0.07561205644472795,0.1129141609581449,0.5644622471201154,0.01981600304413162,0.03313181678875123,0.1469265672177883,0.2282414816697892,0.03245961503683582,0.04204114901676968,0.4554373866427902,0.007445506964837825,0.0477579558665764,0.1009994770038369,0.03678792480688239,0.6578620898078757,0.04346935063842194,0.6407071509237101,0.1707421529178552,0.1073746849146802,0.2683159682198096,0.10036832692938,0.01966661431741193,0.1144587394637723,0.0001854569874974727,0.000238360585131171,0.01680425807850864,0.7191641558589965,0.2158180408004891,0.2912834046176644,0.08924579098542253,0.0289729850859716,0.06007273401000507,0.9202289188909326,0.01877675010860675,0.01499383336619971,0.0269643202659728,0.0559078588818557,0.1370911487324316,0.05626930412369449,0.02430087625401216,0.002768905652399918,0.09419124278380822,0.0003353905129758608,0.0004586139596230963,0.009090550771264683,0.7870245771545262,0.1310579190209814,0.03302722207474077,0.7265757853438528,0.1896239032793562,0.4403402368282422,0.7467078074997224,0.765730458141459,1.508122178558443e-06,0.4663218926620199,0.5047898179046633,0.9251769725459272,0.5400539436492904,0.04498111859285444,0.003583647474984361,0.03314611334471342,0.3655393200830152,0.01756746934354117,0.7080115842840516,0.8103612630955417,0.09747728795117137,0.537745600961298,0.4823448715525985,0.8076398120860129,0.04162857604309839,2.434578856502639e-07,0.06030926080142889,0.7150912892143891,0.8294954916541089,0.03681248102128176,0.744117418131414,0.8763313836932529,0.4742622567827278,0.103923514537761,0.002622606260722494,0.0002468177284081474,3.832413561558955e-05,0.3379569269134389,0.001287540358396004,0.887539806619089,0.0007547060153968312,0.645792138833635,0.4157721026495469,0.9824103838210185,0.09034424937609385,0.3537310517290381,0.001803061679689454,0.2590353445481645,0.9664467650620417,0.9076335788180985,0.5741041593356259,0.002827523480719379,1.008581707827744e-05,0.000541642272780131,5.982719418632379e-05,0.6345206647050011,0.7742464447870098,0.5280420183140433,2.131802338795924e-08,0.1988004248607246,0.9308785993589326,0.792208610476172,0.000480985911688747,0.3876815260444232,0.3419021219876116,0.8430140455781661,0.03221337099609316,0.0003509848052643941,0.07000559042134095,0.8667863331920951,0.7058804036212849,2.183614802619535e-09,0.7323860466692993,0.615338035733997,0.1350686931452631,0.2875212611144068,0.000203471480882368,0.001022064378687996,0.0001138378421935584,0.583377942915583,0.5592455683449866,0.9034316623208543,0.0004853950036418947,0.06004494455168718,0.03891613853787958,0.8483309320977116,0.9996400915293904,0.03497741552024178,0.7687312313817544,0.1707421529178552,0.1808310898366843,0.000744415649473846,0.0870881032849227,0.06001079526042077,0.0002324657924152045,0.2637020366721073,0.6971943353981389,0.0969656660181327,0.003127381586552328,0.3822437158387152,1.491950764356809e-06,0.05744788213448248,0.02307458462756344,0.6407071509237101,0.007786718689054449,0.003207383460240539,0.9202193738253064,0.9995587242417702,9.094482569834947e-05,0.001725534461036224,0.4470019686436519,0.3055694448239344,0.5820155377681526,0.01918543313870086,0.4044215386639683,0.2742888933752569,1.007545446732182e-06,0.6125385149273709,0.8205516754491651,0.4308743360142436,0.7662222849825863,0.1177258572800149,0.615359909322637,0.1212333870811561,0.7101373987821097,1.571887203771521e-05,0.8443400754424556,0.1883770297765186,0.0002998362421897664,0.08795245287982151,0.9577315007099367,0.0004240571636364136,0.06992123481299299,0.2227233194762799,0.1858630730233557,0.9474643564167003,0.8898325148952866,0.9048725207805804,0.6462023246138409,0.5741737211043234,0.2915633155357546,0.001091162580844866,0.8755589837667178,0.01366689744702677,0.1813845615302615,0.6507904977973199,0.0004742105724055378,0.0003280080454457973,0.9296162245967607,0.0003025967497319057,0.9852794336439866,1.665912583231789e-06,0.3671025849389755,0.001857526730966455,0.31494390602601,0.916286011144946,0.02271863997298123,0.5196883816921671,0.006770124778891353,0.001288338614755316,0.9282757403679216,0.3495508575292174,0.01502348542132171,0.01670881440592835,0.01427204650314575,0.1810864901816812,0.2839976107122642,0.124726721831177,0.1178488567500464,5.212268690065962e-05,0.1283650144110286,0.04592975622056553,0.8919277655371299,0.06905144787489829,8.820837890754535e-05,0.1879599126253687,0.6640334098086184,0.3638938010761111,0.1267352864213612,0.003852158937556853,0.3640267310340575,0.4742622567827278,0.6791002409231841,0.05325346597105405,0.01735686767191923,0.9795889899768241,3.589655421921632e-08,0.4837730826714317,0.9227319175224815,3.787245731663875e-07,0.4136890628558975,0.4970982709230296,0.7820993290376268,0.8309675909325682,0.08404425093394136,0.706417618146302,0.9098522555099597,0.06377078622555866,0.002903780997150368,0.8816007973550114,0.0001832608419544925,5.32662279090987e-08,2.507611568521586e-22,0.1533135413503217,0.8394836914287781,0.7024771683785989,0.1652008767006771,0.599372064231777,0.7689539602149791,2.575334744751198e-06,0.01190658913908252,0.9083737472685314,0.2702248499105179,0.02471848736011919,0.3675669666834303,0.7772542237249117,0.02844284156269106,0.01241235671890207,4.272094058794971e-07,0.04525058664819044,0.2616297466857843,0.4851666739502515,0.02508388611835065,0.7256109142191152,0.661995536050469,0.4917739717359544,0.001759592660570554,0.03146619090561877,0.6639625628978463,2.718450628747001e-05,0.132897163254706,0.0003532058938161814,0.4920546652046159,0.8846104391905185,0.6681761326770439,0.4420277735436656,0.04584390932454496,0.034789532257222,0.06603401734008117,0.2647006327937778,0.202705922234134,0.0517457866153256,0.1855154204340063,0.3339410756085896,0.1030758974795735,0.7742147265460743,0.6461588959605264,0.9368770779396367,0.006116036933792431,0.6505304440296339,0.003467978472591661,0.0002345939430378543,0.02244725334236691,0.8786547476026786,1.650051705480272e-06,0.01306137336283028,0.1792852873522053,0.002308773754617828,0.08252385629283858,0.4913420629736113,0.3393458338674333,0.726829551498742,0.06114188460783232,0.9553308424446226,0.8215854340734368,0.2442085879098085,0.1554913117922191,0.01198996775806779,0.4001557657242756,0.002103129207788701,0.01501185208049084,0.207490636052538,0.1786493555455754,0.1869383734329366,0.3472210047759364,0.1478903030839804,0.4811748699356387,0.3148849365951662,0.4974141146746399,0.1485402506193045,0.5818628509901644,0.3622086043007523,0.4959893082401608,0.3341438173294408,0.0002959790880146499,3.042527583539181e-06,0.6804202073420909,0.8795907222841584,0.0003552032569062588,0.6424047600811252,0.560601584416616,0.0003020733355586088,0.7594987774691228,1.579656004709516e-06,0.495379820723971,0.6924809684482993,0.007002424831290665,0.4292044114565884,0.966221862003011,0.5510495969831998,0.9034137997304608,0.1886905233960743,0.4001557657242756,4.441520853955009e-07,0.2210518794744985,4.119154193328581e-05,0.6930862657153102,0.4807374071541434,0.8759663441900337,0.7797782609077046,0.9674102618464484,0.3337968714890088,0.1943556776471635,0.1164880900099969,0.02803347200816376,0.04201129762212943,0.5446739539916654,0.1297006164377255,0.0463704770990073,0.1019337214739586,0.1410731246482471,0.1451666122240391,0.8909550933444615,0.1639548356355547,0.03977992365525656,0.7377773468423465,0.4052706763817623,0.6408172828474565,0.2246687077468507,0.6284622700524307,0.8846104391905185,0.005553595774942795,0.0274837573932725,0.9953662136556667,0.8910399672511812,0.006720628512003459,0.1776023829884791,0.01475977402283893,0.0001230890238763284,0.2719757606308084,0.01789284070032659,0.004578734953947177,0.0008138939434445339,0.001007406904531835,0.004822946384508472,0.004484050334020323,2.04139136276231e-05,0.2370113614299572,0.2533403156194551,0.0002619487730341289,0.1042379016071077,5.768773878618707e-08,0.0001874912592645171,0.2736064075248298,0.4474383922613809,0.1868574268258571,0.4245644849051275,0.5668138594795655,0.002693580755493397,0.0006866216372027049,1.52058666260669e-10,0.645999977929078,0.4507775531568385,0.384164518039348,0.2494544361725461,0.5874160467539318,0.9477615923332171,1.406882655554645e-06,0.7240795006649889,0.5396109859119631,0.4446769186077798,0.402526701530686,0.2731381837385152,0.005046047749179234,0.7918914374082865,0.7268714448097076,0.5355757898521635,0.8513414482102312,0.001762895080668011,0.006758758120773146,0.580092546011696,0.3848333277545393,0.04032725523079084,0.6279536236026422,0.01846092115962566,0.09082242401721259,0.5211721687288001,0.8992379742616347,0.7680962145636024,0.1139174036482999,0.4575995453073931,0.04375157597661909,0.3904642907452636,0.9137989299591078,0.03857031965286204,0.01073051058192566,0.0281307450237047,0.9937226776302859,0.2131192080559015,0.6843563612410346,0.3425215148503385,6.81963133198243e-06,4.012943103280919e-07,0.101387933606094,0.6408172828474565,0.4421201983659218,8.223982339947887e-06,0.01705473499459906,0.06833466548974834,0.8914836082163116,0.004042980442234153,0.0004059490841579003,0.05323895472105516,0.1256565077277235,0.01211961687611728,1.567312676682378e-05,0.2621050376756763,0.03643552399941297,0.3670596185558684,0.356130603615625,0.7524196837813943,0.4593410357649612,0.6300696892107486,6.543594631482087e-05,0.0002048096394673483,0.04169850154971697,0.3121310706702751,0.0006910794842488056,0.5381536798233247,0.2833981293558123,8.600013855402308e-06,0.4290909753325429,1.044220723465243e-08,0.006511438459063402,0.4858753854164007,0.006690715670618616,0.1959405589659181,0.1288532515845954,3.099262904026989e-08,4.872925227655334e-07,0.007982804961038056,0.9560064245738843,8.032001700538973e-06,0.0002755884323388363,0.02517979732930081,0.368587755507823,0.1526170321755389,0.1392747446257326,0.001927461510039959,1.523927897430513e-08,0.6279536236026422,1.001712092225788e-07,0.1662796159944002,0.5773555411398608,0.5114698975297317,0.9869364783357176,0.01169850694883784,0.2212947605289362,0.1300427469025698,3.314967819270614e-07,0.4397189911104868,0.003750700770603787,0.9266761831710205,0.02681050852856533,3.436282171953506e-09,0.1800165230217671,0.9003188779901801,0.3534923354429504,2.916429263903701e-06,0.967160322054779,0.8401324407154585,0.9732694137993985,2.728247229703856e-05,0.178797165987777,0.2837071475022145,0.6929982907774326,0.00668051355824674,0.1368524203737582,0.3746289456233961,0.001063637287354228,0.9646763390698612,0.007357751084570647,0.7755838763495531,0.06920264489400708,9.972399053418938e-06,0.8194115009614099,3.062204449666909e-08,0.2067048897072985,0.4789639021451162,0.8137420783296719,0.1223754803183539,0.9107085811692675,0.7107611341235406,0.0004771127684550987,0.001329340879404686,0.8913118275842538,0.0153085840894762,3.403856794509863e-07,0.2195801491294422,0.3812677956344574,0.6392661683531082,0.3722062686295743,0.01528748047233564,0.3397004709516482,0.6639015632948181,0.6133387779779507,0.2749873264692645,0.02554525769442423,0.0009743410256905558,0.09744901165266286,0.1953799389831348,0.8454296941263695,0.1449075720655971,0.1245670579579854,0.4740097903272535,0.2001791429199266,0.0009962330328221808,5.788950962179733e-06,0.7189478908286547,0.01578856228781265,0.06004170635026156,0.6092840797594876,0.003660189722311412,0.2431999758895698,0.03891613853787958,0.006713668387184423,0.2460671698101468,0.5708006210649867,0.7745152340217568,0.5191444728799869,0.00394950653310679,0.001142503400147824,0.3674456483993552,0.0004136696063333957,0.8919277655371299,0.09508023568121537,0.3422762069598264,0.08098983032534254,0.6945124666231902,0.8198809317728205,0.5363537773665547,0.1774261526863714,0.1177686119654706,0.9308309839185359,0.2690987818775205,0.4000053623921056,0.006704173184392406,0.01304920249595933,0.4327929161475383,0.04728150608507252,5.020119322642078e-15,0.6590773652780622,0.3041732582767491,0.0008433156373266047,0.01077649370901862,0.466929269553663,0.02833783316018688,1.609789484189182e-05,0.5381536798233247,0.1347052896028259,0.03269115627837376,0.8439947762371545,0.5521345203348123,0.3943534580878627,0.5703162423176362,0.001626288732832932,0.003539989000976861,0.1937524520537358,0.01166405098916082,0.001176926514231853,0.270385225249395,0.0643735023188455,0.2745133054664109,0.5013301885486153,0.0001166668372881744,0.002450189125101581,0.001945982101418744,0.006690715670618616,0.0003428145301665442,3.146774884826216e-16,0.7181735493975816,0.001036624789459433,0.3077629976993694,0.7195364390052706,0.002466214215303624,0.8200546315608412,0.8667863331920951,0.7043570216934989,0.6085069557650185,0.7374552293878562,0.006307308202225725,0.237433059754517,0.3400849900907765,0.0001940149668151012,1.659002891065771e-06,0.38644517542041,0.1688581483127194,0.2457311569452358,0.8374670385384224,0.1735647011164842,0.0001454845167379203,0.8967119772062295,0.0008008294620691388,0.05106012259946791,0.179953005306645,0.08252385629283858,0.0003458037674307114,0.05025072779147696,0.08856453217545261,0.6397000488295504,0.003345758950067881,0.7715267148079097,0.2023990561070033,0.4643506191926593,0.7254024994802248,0.0003084523001240275,0.05611336186568548,0.3368653898630334,6.021846931844022e-05,0.3103012204103309,0.1131042935414065,0.8521605462608293,0.0001033490928438248,0.1009255487129712,0.6421506296212612,0.6917622617218273,0.1562070678712724,0.6912292343040878,0.01628724359675952,0.03316897802820055,0.003855992115979937,0.009180517106402775,0.0003709778517584341,0.7333059055364348,0.773903342964872,0.1174107954143549,0.9414328096155593,0.6552587616820121,0.8714900036831484,0.4882826093745577,0.7711080143288488,0.5164565026604014,0.8834720138954748,0.4407690866359109,0.3687561694093667,0.05904285323364192,0.9894953227737648,0.1848995966275787,0.03312977198292177,0.0001166668372881744,0.001310177042504928,0.03273681139404356,0.0001281829890548522,7.634659960659536e-09,0.01186194949569247,0.7711445151336309,0.441818121923144,0.3517035730821587,0.4195573989658394,0.6203554101174856,3.510844884571223e-06,0.03355549717722242,0.5987834380945589,0.0008032602073348318,0.06225837992248646,0.8071056643528347,0.6246357500878706,0.05304415658782988,0.8843249868668023,0.528156222169084,9.5353638731513e-06,0.1621506532152326,0.5521345203348123,0.2632082190657489,0.925561380345125,0.05015117557690191,0.4682657349537838,0.09490917649597665,0.03908851654998693,0.1420954644672736,0.04550330888203865,0.02203372573391155,0.7745327958319056,0.0003441011092295611,0.001992023329794099,0.8241983674529635,0.165710538843103,0.3822437158387152,0.3740660171332254,0.8905333238543257,0.003457469981180204,0.353919697544578,0.783048378148124,0.6329213199573015,0.3942396255635507,0.9546236692463167,0.3729352078743196,0.04206343641370686,0.113656684686888,0.3746289456233961,0.3423003190133498,0.0001068310674208404,0.4300080645262129,0.7549197904039927,0.6273753322231318,0.1758096740171421,0.06239261787090515,0.003055970587376736,0.03099682116340041,0.7737001754775076,0.586715530622847,0.8782789708267189,0.1644419118977993,0.4537828237020667,0.1277779579625795,0.1488330246340403,0.4733359658766911,0.6290888415512228,0.2204478191409309,0.1997909207306224,0.7739831541730282,0.8114813892024398,0.3112920039748925,0.8489651695356301,0.5992402227836143,4.664414622225118e-08,0.04845922590120486,0.1867421749599388,0.009548742025700566,0.01536423945761,0.4593410357649612,0.03123610921140961,0.822710270261539,0.1868574268258571,0.1795887071416981,0.001171444717267949,3.833201880595221e-05,0.07162142654879741,0.1149720055481498,0.1319179874383725,0.07775280659276744,0.2831244807679312,0.748624003257191,2.993160189613515e-05,0.00964047069788833,0.9485120584936217,0.0307611162769808,0.8173337776858216,0.7084333660691627,0.02758920131839771,0.6420160155496071,0.0557262019983849,0.4950198612201459,0.9901999461317573,0.5528223727120151,0.1236090904340566,0.307984787178276,0.2382333972092383,0.6498399416086346,0.3176769946103398,0.8709233908305102,0.04817555020326942,7.781711953024751e-05,0.0001337742515446923,0.7634316299970078,0.9934511459824709,0.306351619283481,0.3596300192620709,0.1380590874748095,0.9824915843506197,0.239579273445857,1.656241135314305e-07,0.743143601482695,0.001417708759507125,0.6788351428133287,0.01702743347501342,0.9648598480122718,0.7839756112801453,0.9844489058736869,0.8839344219496461,0.0004034420979930709,0.7615360125906849,0.1709736465866857,0.0002485057457671987,0.4569338802958509,0.00534849688222408,0.6920340880140179,0.02944787788025673,0.1206926546126804,0.0001835014729351654,0.001095496403373738,0.2603084255762309,0.4469085971434634,0.0001020378064062736,0.001431550814700525,0.7203530045103602,0.2779861404118119,0.9275369685350772,6.990390297712733e-08,0.1077845308488,0.01398030224189223,0.0446335016923974,0.2739270921305027,9.069686874890054e-07,0.9907369195363035,0.8198052690759815,0.2941231351606834,0.3941456306026629,0.003750700770603787,0.0001222233396809298,0.3074974770720146,3.116486453987323e-05,0.001992491780814443,0.0127074279954301,0.002236718285357346,6.569351315001811e-07,0.007500666419494252,0.05163503599481085,5.600388236417096e-07,0.0282536645000618,0.01654851691315359,0.8601224063508346,0.02555638910892928,6.307911663354428e-05,0.1382996840471104,0.00656359178857708,0.04592044264058315,0.04002373256166107,0.5147286543804835,0.6802479151027938,0.06745358431868427,3.909092330470305e-07,0.3293655392152437,0.01936478421687373,0.6218333655152144,0.002772456340056832,0.04359151949709654,0.002889887638803651,0.002554947278840609,0.2192380278476578,0.8417671936750322,0.05981292440951171,0.008500607287752123,0.1031274811843759,0.3584052331542366,0.8692619594240978,0.004836767401469685,0.2161646094460251,0.2775165801323667,0.07566177936062116,0.8367152827676988,0.6783516513022223,0.7173547010867788,0.6564839853632175,0.6912292343040878,0.9474643564167003,0.0006711225281884849,0.6425763855793343,0.9882719404698929,1.672200927720219e-05,0.3365975883509396,0.5746401193926497,0.341964106776812,0.4568137471072723,0.7318732816588687,0.4576950703038906,0.571654509401344,0.9952594379297782,0.1175290593059779,0.001959603828550852,0.03825796676602171,0.4955386718590295,0.6498399416086346,0.378950655947527,0.3844348037935529,0.6781899423048977,0.4710400190176429,0.0006233270556811438,3.410700077415409e-06,0.8017107733394531,0.001066813704771509,0.4713508812069368,0.1731819733613915,0.3593863280881636,0.3890115530465846,2.986900475797136e-05,0.705273127611212,0.5349433942498284,0.5909324557836838,4.133342689160157e-05,0.2089383026952482,0.8825794224565393,0.1000215356843811,0.03013193979819053,0.05278627350249183,0.1373178515928726,0.3303823453111422,0.004057909245011869,0.6838266233184714,0.6695902308065332,0.7909038092322691,0.01624982421332856,9.269492212441354e-10,0.7412446019220612,0.5049874863295701,0.0269643202659728,0.02057856261065683,0.5260820179431205,0.8696042275903381,0.2136287934453245,1.647806365181892e-06,0.004420819823798191,0.9651602873872321,0.02151481457031533,0.5520507054743581,0.7867340190814948,0.1320286113704765,0.004527471009736627,3.59852333680009e-08,0.1295576268248302,0.04283634310450432,0.0142983829293325,0.4418608086869105,0.0002776918299324638,0.8003575975907392,0.002066863172417209,0.8972735199531928,0.6720888505599681,0.1428019428674621,0.001449923809236388,0.0005790024751824105,0.002579281061954985,0.1572518302632196,6.766391944762663e-05,0.2800849019477664,0.4271229537642844,0.005995182773622838,0.5378650401978778,0.7815576867706236,1.146351337481142e-05,0.4690411666352498,0.9139414327780961,4.127238041429685e-08,0.1368225555610098,0.0005612431791082401,0.2708706725411095,5.364953087801921e-05,0.5392649870733451,0.9730861266457792,0.8048275893137194,0.6246357500878706,2.606810960652912e-07,0.003280552644814414,0.3770557597676538,0.877604482057851,1.863356466115634e-08,0.2857156289549785,0.2806614574434332,0.00011872251363534,0.02056239198791225,0.5521791565669338,0.4346256252842931,0.4499742942652868,0.3624813870965586,0.8327670310797102,0.8982176716853583,0.9210329328674836,0.9354646124900917,0.5960392313497541,3.610841229462031e-05,0.08764310086358687,0.323692673967767,0.1248304950183346,0.06753894925208281,0.6162893456390677,0.1155829320342454,0.7738352152207233,0.8595283871742047,6.74849474219571e-06,5.138032268292922e-05,0.001095496403373738,0.7467078074997224,0.00605739234120362,0.0291029373828673,0.01432211170510217,0.01800044221619908,0.8309320068543062,0.5922824561934159,0.6012050225780471,0.001650596242938279,0.1444881824736701,0.09566108937122267,2.850868883026127e-06,0.0005758337524138729,0.02085735674344706,0.06127803159970005,0.7304271623087391,0.03113806119559312,0.6926157488206697,3.306430373842624e-06,0.5887725127913946,0.5315800531774758,0.6684315176564467,0.186856777386939,0.6408362028484291,0.01578856228781265,0.004742398282503825,0.7698198411563923,0.1058446451992179,0.008935823861893783,9.069686874890054e-07,0.04066484356568172,0.2396964275499578,0.3360553601094302,0.7375397871367821,0.09876223338805783,8.105083769964792e-08,0.114309637676076,0.5047898179046633,0.3332533381388386,0.800622510297723,0.8205516754491651,0.0001218425504073503,0.9766640278465979,0.6274201841919975,0.209892948122852,0.4574444797455093,0.1013718710992879,0.2587447292404797,0.005108572345095329,0.03633733803253866,0.3122535313737901,0.3251917465936393,0.80383763986973,0.285660413701627,0.01075195076459354,0.3051662992780267,0.2282269055351131,0.1256962329988703,0.0006754101695695677,0.8394108513847029,0.03418812696817061,0.8655732869387736,0.0421708321575214,0.155834197758046,7.113417269561352e-06,0.6699408684527994,0.001645243777643688,0.05762260875842402,0.6849766476832646,0.3383737170467864,0.05123863714199869,9.968937938512719e-06,0.5808159184537018,0.8007497714694602,0.7797782609077046,9.151177896764955e-06,0.1405112405876211,0.8177428375370043,0.1511641677291853,0.2675953985814183,0.05008914134364977,0.05263213473818302,0.08033377365844807,0.003961887047869313,0.08418536845222724,0.01886084339992112,0.06651053301358641,0.004115823916328719,0.03695308813835332,0.03244288922910973,0.5847448001638519,0.05194868686598756,0.5355757898521635,1.227531641505677e-13,0.4314998127353725,0.9144283575549412,0.007832629275263157,0.5960069960258688,0.1320286113704765,0.06800713231678443,0.399517507517526,0.3347908322928776,0.2072800448187038,0.05348029554114125,0.003961773700616681,0.3631572644485753,0.001506787712465661,0.009622378706941903,0.5970262983300291,0.01290891241646519,0.5438293970391275,0.1039578289710231,0.1532272500412543,0.8467475180631973,0.0006767139663238592,0.991441096677852,0.8846650052665372,2.718450628747001e-05,0.661877380606845,0.8005802367205138,0.1707349264169115,6.330476145246938e-07,0.05649275334681458,0.004600096865247609,0.1265157332665174,0.3081476243870001,0.02131434025244461,0.05342903091957823,0.9746384985077886,0.638228012475338,6.602827736105784e-07,1.270422540297586e-05,0.2662320083312664,0.2768834491069528,0.0003529086757283233,0.2881548912783071,0.159185763124334,0.4078352652560524,0.5719894060635703,0.7103732009841328,0.1114736988818338,0.1236274677489219,0.9563938413518758,0.0003529086757283233,0.09715952949709043,0.6547866667708424,0.1221984860105005,0.6485031166108864,0.5728484855246055,0.3701975030958125,0.01783531242896416,0.891131492131317,0.2776541083011482,9.998647403253479e-20,0.1912792301196026,0.7866646725478915,0.7472272634964271,0.0005164709228285755,6.628461926247555e-05,0.01644196026024882,0.1110572510099106,0.1318790906764331,0.5359707954413061,0.02878145057003234,0.07646817994234822,0.1582685485625607,0.9704624961230964,0.4368656646261094,0.3335248222359991,0.1399741834231116,0.3741712948046189,5.262616251352779e-07,0.7393123182373929,0.7343530251695406,0.8633801235724239,7.662850306041934e-10,0.7355392519341134,0.6839625408196878,0.1476257501865936,0.2406353100505265,0.0007433408921489648,0.1339342769014724,0.8084403921553645,0.55313648042341,0.425131021945224,0.9781529615324478,0.6464515763247902,0.000622519038621753,0.001990143575529482,0.8118030840288395,0.74227189985839,0.003354479668780276,0.02445382970670921,0.0567792036970548,0.9100610035416831,0.05909692334742008,0.002932549150790958,1.257736631414169e-09,0.7685965262254677,0.3328487789259834,0.03336378350588715,0.2279033320650283,0.0001652068311936367,0.1340813508218436,0.5134612782658727,1.523927897430513e-08,0.7362528578975966,0.01616177400169869,9.246873064239298e-05,4.37108683211438e-08,0.3555593491846245,0.4247255720819451,0.9836720990572133,0.05702088640770474,0.004970485756260739,0.1486482043874783,0.7630092975182909,0.9863452387947657,0.2591055893144781,0.6277636481359601,3.765669629541131e-16,0.1277779579625795,0.001670689289736213,0.03166338784387055,0.01512924431475904,0.1729693395243265,0.958434954909163,3.131417838053483e-06,0.8004536492826924,0.3454412484850428,0.6404009288654757,0.7417775491710438,3.717979894078627e-10,0.4892764663388089,0.4811748699356387,1.067599123276048e-05,1.306437531953355e-08,0.9202289188909326,0.005307586177546813,0.4476700928529679,0.03221337099609316,0.0006289440371938126,0.8518302758896724,5.525274248011558e-07,0.07008264728834956,0.5529396979120712,0.03673527619751767,0.6936349938096997,0.03335139335948647,0.2577145436742577,2.753480390532461e-16,0.1649244489379334,0.01529447191505239,0.07968697510940675,0.4651688205265315,0.01603831261882827,0.2700484022080095,0.004660273348386939,0.5482236195762433,0.1348896075820903,2.108706837390803e-07,0.0008979432602012307,0.0008888613144014818,0.003685846057393788,0.4751428851829839,0.5312156048355812,0.006532359194146753,0.8639707864103457,0.6133387779779507,1.391972225033376e-05,0.06275629579233515,0.2007007748565381,0.1659607789619248,0.380901162125563,0.08568442966383667,0.7898581695873095,0.6358669646631538,0.02680367781825271,0.06887410745900728,0.9259719874749441,2.796546230714905e-10,0.5503834768623145,0.01492030031178538,0.01751920795323811,0.2367718652735286,2.769429414036324e-06,0.6892961234689523,0.4744480141430499,0.8837214916034932,0.2438366008161,4.383330557885868e-31,0.3042141412819262,0.5691899420181895,0.005318585316958026,0.0127074279954301,0.03870744420740743,0.197340948489201,0.1026970604356443,0.005993609723682175,0.0001934334820779602,0.003030732091944837,0.7797782609077046,0.9635653976132048,0.139603227193667,0.5127828263477596,0.3848826805065352,3.980021939496266e-05,0.0477579558665764,0.4708121425666394,0.5386945957132041,0.8714900036831484,0.008248501045089665,4.313911093755004e-06,0.008341842332236655,1.850988082538802e-09,0.806143716565741,0.1669937149793353,0.1686329830447924,0.01968744402259432,0.03482348991320424,5.111096650596577e-07,0.001335250328797658,0.008743687902755459,0.0004108196852906071,0.2123844650821896,0.1694301448583137,0.07322196108715899,0.5550590877630928,0.001887856553402859,0.3792757018297194,0.9425356132940845,0.3332533381388386,0.1988004248607246,0.9614032520868224,0.6133387779779507,0.3456556492902566,0.9831020147812237,0.009174641179831812,0.3163501129762324,0.3965397534891736,0.1406581518545195,0.4722584366965388,0.02656882575491572,0.1022776385492172,0.009033581581499292,9.983629227622619e-07,0.000219830078570694,0.963664347060495,0.0256063965822492,0.001524116776893743,0.6051326166607509,0.5282958689185843,0.4876646992206556,0.03781274198470644,5.602288201174539e-05,0.1340168686829016,0.002350990978482413,0.0003663992001149855,0.519132942494467,0.6992867330682668,0.1046387617626164,0.01966189872294755,0.5890457804345194,0.7901777693374957,0.01700452890911693,0.3276273462684258,0.4448533426313778,0.3711610510208976,0.304595801208598,0.6531957976286137,0.1907998088401062,7.088808865754061e-07,1.006736377666989e-05,2.014367140446895e-06,0.9004135330176521,0.2512995052651165,0.1606031248812073,0.9171912933885725,0.2143591003854007,0.8938442570527766,0.98034207091734,0.1252836805388372,3.67941588945557e-05,0.002772456340056832,0.340002615345927,0.9453444246908607,0.2817713156910994,0.6951415137480016,1.121176572823413e-05,0.250141353654873,3.979912950437074e-05,0.7971631751309095,9.115526614404055e-06,0.07598043002945816,0.02430656259479151,8.692357750890447e-12,2.510314210837264e-05,0.8348381942205942,0.002849422389716031,0.8231007849825646,0.7333059055364348,0.4375867726236236,0.006327265804105098,0.6046465065537995,0.5941386269397787,0.007516209691179012,0.06266036604048868,0.01771068783657909,4.129837829804198e-07,0.8616642987357883,0.01167242918432577,0.005233825517076026,0.9854108836701665,0.1950186328069967,0.08404425093394136,0.4266103359126245,0.02623042449122363,0.005162609352909187,0.0002776918299324638,0.7154167343906529,0.9024605209552944,0.5630711639349798,0.2558640848234926,0.7908527548093673,0.5725833966481078,0.3570883574253487,0.216857677936779,0.09430492235062671,0.1079258649907685,0.1031204200769863,0.02037444619073825,0.6215877094541781,0.2536755400053454,0.9369282095806543,0.538193880039897,0.2982772234899038,0.1485402506193045,0.1827392873338616,1.159393634422693e-05,7.672935105712641e-05,0.9983541428540438,0.04859233120246047,3.190830572995348e-05,0.2583833061198132,0.1085874134950669,0.4373020846022043,0.624229056577327,0.05559483503126791,0.6589280056993896,4.06504550965072e-05,0.5091444304688676,0.3658297372392355,0.0001605076449805925,0.526678959732931,0.1035029253714052,0.8393561668968763,1.622002867468635e-16,0.04760643978166102,0.3822437158387152,0.01198996775806779,0.2821566895196074,0.8397818612856659,0.02295502793664026,0.04835881830385124,0.09240176360431776,0.7209613042191755,0.0004057412490065139,0.1221948834728032,0.8190632143720433,0.02082790707227924,0.1141169167050309,5.574809342242923e-05,1.248403635253704e-06,3.706610738579053e-10,1.743489523142887e-05,0.4443903297451697,0.7307886336992906,0.0002432230460388119,0.6262822684128501,0.1085033420526935,0.1439056318239433,0.002127557567493314,6.239671968362459e-05,0.209616384902628,0.6730251330357788,0.2059843540311875,0.4667708626376122,0.07025126295251517,0.143635348511906,0.04525058664819044,0.104856882184438,0.4245644849051275,0.0001256976361765863,0.01320943551552923,0.2632344958028394,0.3608126149729783,0.09557215583438576,0.0444697275348151,0.6278371808528178,0.04463209846712236,0.001775400599786942,0.002099856304975275,0.09690316733939552,0.03245961503683582,1.963333567444154e-06,0.2963016401258595,0.8652664816488063,0.2561373288393596,0.04441421756087027,0.0001090416749458562,0.2412613079864822,0.0006479329590245059,0.1745384452125057,0.05048861853437588,0.2756565277590604,0.9852794336439866,0.2809292279087133,0.4456616759218669,0.9261881631239571,0.01803401426875245,0.01014019885794144,0.0446335016923974,0.1853076348141191,1.040003015478679e-12,0.3398481690126178,0.002408667853081131,0.645792138833635,0.01752076110824682,0.01607145204223555,0.8188129905540558,0.3910049238264954,0.6474538621032377,0.4661192816209251,0.4950981211281192,0.002953052735498981,0.4412262967210333,0.669402382528662,0.06813402439870553,0.01735204190442844,0.004987992837035418,0.8571439569355287,0.006934323015751869,0.002345061583879018,0.07815746204382576,2.507817838987566e-24,0.1240124755901705,0.0004243252098641196,0.4397189911104868,0.7295897138823045,0.01434842139093104,4.984387196051032e-06,0.0003181826972800745,0.1901961233670589,0.6734434660698077,0.008584110162595181,0.4167009477444967,0.01369680905368895,0.2101929180267143,0.8498847235088017,0.1998046195289485,0.001868539901057866,0.002187398353300715,0.8690849992502931,0.004587775008197725,0.637439656817638,0.2112318093615492,0.7254024994802248,0.3674456483993552,0.7826320646318826,0.2525878536067782,0.02382200740215572,0.62133470672206,0.5260820179431205,0.6563436162430564,0.1790547539692473,0.2252633600978533,0.03050554352173116,0.8822961956905553,0.4500307190794612,0.02250037740049012,0.01512877549661106,0.8736766239628974,0.8237502198977877,0.1290943333041645,0.0008458191490708207,1.540028919772771e-05,0.07562192392178117,0.01352083956448791,0.0004067511199538552,0.04449915682050216,0.8839344219496461,0.03831646449068804,0.6564175249748082,0.9700959778860879,0.02704163918010748,4.297999881083412e-05,0.01488842949146306,0.5076218603906194,0.0005422121538120588,0.1013718710992879,0.2064761635630959,0.9047347328000465,0.6389731408085969,3.99572840407603e-06,0.05486182708331373,0.07263043667816815,0.9455738705970125,0.463225132692161,3.449968407663977e-05,3.554599630318966e-08,0.6187124140054189,0.5543363277401123,0.4819916549474967,0.6288900004672411,0.06021318225719276,0.708321520900987,0.1453819937866792,0.2815861144021491,0.2194453904220377,0.8249940156513059,0.9402737378740078,0.0004576845565196022,5.629395039232279e-06,0.0001692012794936793,0.03697880918671324,0.661962502128824,0.3196417883758199,0.03128707478926276,0.09623582264162506,2.153243453300086e-10,0.3479178246293142,0.8293856695632259,0.1405309038177338,0.03776208709232829,4.231029208848721e-06,0.8919277655371299,1.048059592991636e-05,0.001314846593667099,0.009779268556201431,0.08880554246183738,0.5159596914005393,0.159185763124334,0.6543088212089084,1.621286516125355e-06,0.01021319562513505,2.665025614717472e-17,0.8846650052665372,0.02878404116469898,0.8149061726130757,5.221321416074924e-07,0.5431832419984362,2.249336903904133e-06,0.1403646446251332,0.03346529502321031,0.6385060832779694,0.08561943494901936,0.007421929300416425,0.1435012008069153,0.8067752574346339,0.5723458886446896,0.6904459626049985,4.395736144574197e-05,0.05785214442547015,0.01604450421987512,0.4882694657337956,0.2750993785401231,6.602135472652828e-08,0.006964988900611057,0.04978964303165222,0.6054862830208223,0.8783928109525093,0.08092883573301332,0.2642416573043932,0.006680285308890083,0.3653281496956803,0.2202990736618499,0.1200978288166288,0.07073733086959628,0.02596146839087062,0.9617441452006575,0.7720888135500441,0.8348071297559319,0.001448530970782369,0.3966137796918817,0.005860507709575223,0.3149248211579726,0.6681761326770439,0.0008689920355128062,0.01605331873215825,0.117717437952299,0.007899826772762478,0.6038908631915254,0.6929982907774326,0.5495516188241257,1.488359050418319e-05,0.05943633320232407,0.005072467757866558,0.1697567413142327,0.2258730863254758,0.02389236755736205,0.2395602595563478,0.9242514427499066,0.6517786528640718,0.8003575975907392,0.04949377980340548,0.02584815253703613,0.5844071103780565,0.6408172828474565,0.0858970647868391,0.8655743720826593,0.002372096889562409,0.685477989836521,0.001135006489396136,0.009447216629017352,0.9031345570029817,0.09488447238876804,0.05121799688458062,6.61651906093403e-05,0.03873827408990296,0.04179099689343314,4.895274615502365e-09,0.0343343799959688,9.753288210441336e-11,0.4879506785745615,0.4372037677887001,0.1860090195133845,0.1815440394014807,0.0798216634083839,0.0122492342212021,0.003870828089844828,0.2383309380354992,0.4593410357649612,0.2078511628859993,0.01742152179415624,0.4899836701749448,0.005674064975935571,0.5602819076895378,0.0001481015772189013,0.003611425828360824,0.7323860466692993,0.626278339905781,0.09891844672954468,0.8304129322816477,0.2578364428883342,0.9444635942052373,0.3685678458001914,0.3904642907452636,0.709080734269152,0.2330925661340365,0.6264106449397931,0.4226118967170031,0.0002841265018119623,5.03952478614493e-06,0.439747055068124,0.4205708413191743,0.3276158663941455,0.5942406031184002,0.7288317005312693,0.01472805952146931,0.6497213725458773,0.01091123389988809,0.822710270261539,0.3014461319662303,0.9337812056352016,0.0203291091670976,0.2049417051728839,0.695242895103118,0.9039735535263234,0.9188052201809555,0.3035338770311428,0.0129564227103795,0.009859980385668559,0.001311327113679115,0.5726407337823777,0.0001252240499629114,2.743898903229011e-07,0.8752148532844932,0.0004423407915287816,0.004809025649911238,0.0004960522397635407,0.002441032995098976,0.001456374664825281,0.3816410731503998,0.3842655128625787,0.0002921619157894701,0.2788246322039891,0.0031809586999021,0.2194453904220377,0.6085069557650185,0.2996744623872043,0.8718749957514956,0.1092624750569497,0.6425763855793343,0.000676397364724293,0.5460568103294948,0.0008894608240594646,0.1587483439397586,0.9090089726002256,0.9016956275891993,0.7813552650888332,0.00111276066666771,0.03147978639247401,0.9421630639701442,0.4777464406418276,0.009252551245705098,0.2928303990239797,0.2093639905029931,0.6252649693508103,0.1731819733613915,0.6688782945382619,0.02218746709000853,0.6741793113824265,0.2583833061198132,0.7801702015249835,5.573886724356735e-13,0.0001554317614336915,0.3175430992828019,0.6813406227583066,0.0001095983552959098,0.1492597017406687,0.02180354822359041,4.395417810917798e-06,2.739834716341492e-06,0.5198972808492889,7.014095807916599e-05,0.6704484076286662,0.3260190163215867,0.4475552980630479,1.301175535987829e-05,0.0002504127602046336,0.7166630783696201,5.041049542138354e-05,0.09774986260830666,0.1804691871521636,0.1956387027224916,0.3031588232443793,0.7452837179928263,0.2904291851714845,0.1242868261511241,0.232142072583613,0.891562178225777,0.02901479742237415,0.2999478703511758,6.330476145246938e-07,0.7686785892478146,0.310393431784515,7.341039479591051e-06,0.12191851051177,0.5011904279791856,0.9894953227737648,0.9213184440884313,0.4711722489055461,0.65210996202718,0.003677596097890469,1.343090190303132e-05,0.6556189624442008,0.04755154220024749,0.005963256925524874,0.004410292353660348,0.1545683343099179,0.963664347060495,0.01371404456960296,1.66047946715088e-11,0.08139700280982874,0.005233825517076026,0.3116069008182351,0.1164803149699048,0.0001181996629717561,0.1967230072553833,0.3137065164364057,0.1556650044720995,0.02906683853077673,0.7439870040446207,0.4385484648079511,0.6006549947448483,0.7058804036212849,0.1565920785845581,0.001075362620224226,3.156799067469659e-07,0.765730458141459,0.08264873570505972,0.0009530095644540781,0.001959601248230101,0.00504083054300178,4.472782554064998e-08,5.392003322804966e-05,0.0004627764788904682,0.1652008767006771,0.6201780175511776,0.01045665020746539,0.3738589351301622,0.1395385590975705,0.8741767488414119,2.793004240195247e-05,0.3689405428663993,0.05390953363489531,0.1175360664271995,0.40451041499023,0.5185617890942578,0.8909550933444615,0.00625301049901761,0.3084614770411561,0.01883660420238062,0.9607606821186884,0.03791113907632417,0.004670862593310481,0.1310715824786871,1.16055499343865e-06,9.513539211933295e-05,0.2441367835960459,0.5717699314309398,0.01414300827368834,0.231018200748991,0.6367978038119133,0.7727206489769278,1.729015418064647e-05,0.000454295761903989,0.03201890900449889,0.02094416593664142,0.5025695080206248,0.004991275765866232,0.0192496227385969,0.3606123340784995,0.7467078074997224,0.6039436971717954,0.4300793411019845,0.3349756542107239,0.04149914840776495,0.2335909663490726,0.2258730863254758,0.2944160427788134,0.9524919743482566,5.213704456081419e-07,0.7780842429104845,0.002528597518371902,0.2702257439167583,0.001604753011204205,0.9233161040252401,0.09899232418080722,0.07267611448661029,0.9299780832557688,0.3763926114451266,0.8025972564538145,0.4835697349827357,0.9931072231272373,0.01636776077662244,0.01909676749638499,0.3212578667926353,0.4803278632788074,0.1223894163240823,0.5134487672373027,0.0008468080552123955,0.083481494014468,0.002518975191472102,0.05211789744387887,0.09036185958778031,0.2393938098348863,0.008614060291610416,8.55386277534456e-14,0.06222542923063467,0.01601878041152693,0.3129711877400661,2.036807464605475e-06,0.7894776474325458,0.07057084335175902,0.05490936997594985,0.1332768109806729,0.537745600961298,0.1399112393321188,0.5963742865677238,0.4750633881245883,0.002476241544372525,0.8460126507081173,0.7920517171555899,0.0004895132324265727,0.5009667981935684,0.1582414836893647,0.3361408668286799,7.620923138677704e-08,0.1897262264882009,0.8625782843767539,0.2285787438271718,0.3902893573535833,0.9341240776372614,0.04097761400030527,0.4949924305139252,4.16405780740247e-05,7.85428339597975e-06,0.0001151084225417439,0.4659187490991687,0.09706564717028124,0.01769601027675783,0.2352441074929758,0.9295685042466575,0.1714417076973296,0.0002226413061050066,0.1125920272830789,0.5853957555616968,0.4021834747272489,0.5419138318064535,0.7728286304141666,0.2804337502015505,0.2018384691283832,0.01846474455693951,0.2239198791011858,0.06884337901944698,0.7590511471871464,0.0001127757369934149,0.07530388878228357,0.7195364390052706,7.967077331650313e-06,0.2097053213369122,0.006525917783017589,0.1486482043874783,0.1014569962841418,0.2792308502674536,0.003083976394207113,0.1758096740171421,0.5127828263477596,0.3012657214895575,0.02646572696769933,0.2377185983838752,0.2004453792525585,0.0126216740474152,0.4508915719862081,0.432546673984644,0.3163501129762324,0.05236263347181806,0.02794694362489905,0.1337882786465856,0.7836316257446331,0.2140439003904876,0.9462605856800654,0.406951017706053,0.6751323773988521,0.0003020733355586088,0.5679743125733839,0.01006601073560089,0.001171444717267949,0.2489053167905904,0.0002817063980218171,0.0685823009061293,0.006261545105715405,0.368564177072134,0.8902792200815426,0.0067333524763417,0.6478448062432947,0.0002468177284081474,0.02183465642229989,0.0001127757369934149,0.346692480558943,0.1954310892736873,0.01862085572714034,0.909984737981573,0.5035031374150872,0.787304168617181,0.0001032579626659696,0.004354055545212241,0.9860916844745385,4.602247370696548e-05,0.7316090380324349,0.705273127611212,0.09721035982319146,2.53140796300287e-06,0.02968333689709232,0.5123996749722957,0.6628707849167981,0.1978983495348146,0.04064797583275678,0.9107085811692675,0.0003767060975007875,0.1628111031112459,0.03128707478926276,0.4274474729885787,0.06039925031148648,0.2004453792525585,0.4619924132013368,0.01768848760224202,0.5380250646583077,0.000676397364724293,0.5717556283400875,0.9558150584745628,0.02704359121408879,0.002150528231203814,0.09884755052556152,0.7867340190814948,8.374818736103802e-05,0.8542649232605477,0.563654875639284,0.1119395594567396,0.04947122068902141,0.8962812949021697,0.3615675653639938,0.6093363220427346,0.00569084459921002,0.2985825678890999,0.5417844250867309,0.2175922529985906,0.1571979754512086,0.7230175673145156,0.8898142649496492,0.2513586406402301,0.8448825728047383,0.07930612440781605,0.6965539129402493,0.06041311458330729,0.2098079965167439,0.5222163782181989,0.06997669013833978,0.2876631198835086,2.535579028447971e-05,0.2953856886417403,0.5037205149999638,0.8226723760557845,0.3323659914667785,0.9799604837772428,0.6085018189757541,0.01432211170510217,0.8786547476026786,1.369434044615522e-05,0.5051343479949396,0.0003830028439955747,0.7118249260764762,0.248842582297686,0.9241599531518112,0.2148013167484724,0.02422989031596563,0.275497378290589,0.2380481798755452,0.5121003998523723,0.07079654831763545,0.01319426608061069,0.04391062989895161,0.5090896694415319,0.0590204762506843,0.0003529086757283233,0.8155128190913972,1.127228702453496e-05,0.07097043872331162,0.02100037409168266,0.04989057870135073,0.02951407018545786,0.2527619069945157,0.9016956275891993,0.04066989833489263,0.5510495969831998,0.0003396686509400806,0.2242763066577539,4.007489956347491e-05,0.3946826669058223,0.005220045812299173,0.6583795609182083,0.1394469963718523,0.002103129207788701,0.001987402074645007,0.3608126149729783,0.3354496466008202,0.5139746536968679,2.949408240893464e-09,0.8981768256616506,3.52057624190216e-06,0.2169817655726306,0.001925421759440135,6.368167305250637e-09,2.05469148404493e-05,8.504755405412216e-05,0.5246023052305091,0.159185763124334,0.9939109395728927,0.2224753314196571,0.2900977484204964,0.5069872952671683,0.0001240447836330064,0.04368685894353037,9.680549188474556e-05,0.9556891409109302,0.1107646214155589,6.994314820487375e-08,0.03971951470029312,0.004856792444870603,0.6788351428133287,0.1289986272787107,0.001689578513841581,0.4984549709197741,0.2441367835960459,0.0003187257586732323,0.06910677479991982,0.06042247740061827,0.6639625628978463,0.0005283363151134183,0.00384755005996189,0.1947610640601535,0.001142503400147824,0.9554556796586119,0.0001722184868778277,0.0365311874203555,0.8568890525836043,0.7779487974718282,0.8370957352427816,0.2347151833867424,0.7093769175373703,9.041665489360211e-08,0.492890337921527,0.2750805263766428,0.1583603790072541,0.6764007425132146,0.8657840602129084,1.553128958003194e-06,0.9600929997694408,0.001080422624267077,0.0001221979473966235,0.06676500431647861,0.7994686268630207,0.3067656878488694,0.02897328695971444,0.09824180898375279,0.9216119001427022,0.2639419806555073,0.5037330436573185,0.1203388875918189,0.9218974238774247,0.001797608789433538,0.1483822979350211,0.1239235272537692,0.005085826763973279,0.1803656306191921,0.004264587171236475,0.1060828615533371,0.04354111616173434,0.2307070312459777,0.1009469824535854,0.0008053608114082507,0.9639058533246051,0.4040321162379268,0.4132460852655823,0.02715249614770799,0.002244783831537209,0.175915591983449,0.4750633881245883,2.534839707901582e-06,0.5203371102417594,0.008618308651575179,0.002979267739679177,0.001109012429035858,0.02318024768138083,0.279543950479926,0.001329340879404686,0.7534663889935825,0.06722054667174392,0.9909129887679112,0.4397189911104868,0.2145007300877602,0.8369783136067924,0.439747055068124,0.2666143268161918,0.006628323979929631,0.7892415567049912,0.0001496282458969321,0.4084187410573644,0.9612622991963727,0.1881023181456024,0.1191834007211347,0.03647958321080837,0.08122469862896918,0.7528692218153731,1.007460467785196e-08,0.8182550504765136,0.1654138427341489,0.03669544384306649,0.7261114955102597,0.211614849091964,0.001372915934452553,0.0653646066922995,8.766352961251161e-06,0.00633032836963697,0.0001892044656934951,0.02437860396578188,0.006329488649152026,0.4884800210253348,0.04643963756397914,0.01795579403663874,0.01410493180726692,0.6167427945919683,0.02776916073342121,0.7093809139099545,0.9188364795036531,0.6914845800927206,0.02865572857850524,0.2221865583359649,0.09727034297904388,0.4523759289434667,0.1015182147024308,0.0002882100874717943,0.2565260287285688,0.5386945957132041,0.9879403394768415,0.032532222202497,0.647517514237166,0.008885630604408196,0.3671327717306888,0.003349224190106358,0.5281045460864051,0.1374977833942484,0.01551188157029444,0.01574154856782932,0.6993399676533921,0.3984356330344446,0.07299087281873494,2.482357950197492e-05,0.9686618370976594,0.792208610476172,0.9579682371170342,0.5542963600044858,0.6421506296212612,0.2063952318856511,0.8460126507081173,0.7352293498029532,0.2048345337496913,0.3903301551423223,0.1041616005518773,0.007593000343578286,0.0005869954749934206,0.8247474132664214,0.7356718310697026,0.5441146934667176,0.9877909072459107,0.6421506296212612,0.008788473510535195,0.6741793113824265,0.105625378557279,0.4609278934603216,0.01680425807850864,0.335659953693635,6.845081322824408e-06,6.270399032496997e-06,0.3698318718186109,0.9201323859561454,0.0002505736869913943,0.006395071964426731,0.5819864909633919,0.01426202531532646,0.01171760677996835,0.5741041593356259,0.1889637985015461,0.8278787912879401,0.006589552870215154,0.9918033650321997,0.319445152345717,0.8564610357247064,0.01114730316963855,1.830136652665371e-06,0.2775165801323667,0.9510177664294567,0.006490739196242616,0.9519322138166636,0.008443585597199146,0.5140187892822086,0.3823763398975853,0.3383737170467864,0.01741175133133131,0.7570514425346156,0.001011012063592681,2.026483067150836e-09,0.02022240040109267,0.02022068601059342,0.121070384917068,0.7143165468678661,0.05850054683031197,0.002579907786712969,0.5474757706971899,0.4626505124848254,0.1145484828431196,0.08404425093394136,0.9170754636300545,0.0002943525313705621,0.08963946887231269,0.001422858036999679,0.06306128912543529,0.1960914753114646,0.3308848139810942,0.00666913306774852,0.3084614770411561,0.0001917159004014553,0.4383282910611496,0.4199136390171966,0.7717097827887253,1.108326104334633e-06,0.5618063946485908,0.4496688709850588,0.01281837859036808,0.00602304912951908,0.7333059055364348,0.6266922275582824,0.09641431597296235,0.8188129905540558,0.1186658738932243,0.3221145906521765,1.263044636077157e-09,0.7443828604618512,0.602620763071661,0.8919717428562399,0.0008893540514233158,0.002538213942839527,0.1931568190037343,0.4957979018309117,0.2845758064470607,0.01198104690033338,0.2441367835960459,0.0002669350601527114,0.2053750324036877,0.6447659665917317,0.00509907666468963,0.07988071727740198,0.7829692509764705,0.7549197904039927,0.03142955120548765,0.002760694122013438,0.01206447643892877,0.4232712938712582,0.644462663852934,0.4881738409708696,0.005564283302170745,0.2234183327229523,0.1578969655725683,0.00967859225201091,0.02064154733252905,0.9581182606845825,0.1239471745250098,0.8332937900089283,0.004421900428198844,0.5717395465971555,0.3315376744671479,0.03306803506406455,0.8100670983847638,0.03341788579866835,0.3887366352748619,1.369758100906575e-06,9.543297966180908e-05,0.1976881456443549,0.9275369685350772,0.1181245346782153,1.58425283446224e-10,0.3162492013114846,0.3584052331542366,0.3220010680525731,0.7535787764301751,0.8849671031508964,0.1325112686536013,0.1672570040156478,0.4630967513917857,0.02521265325563951,0.02640458443545643,0.6101978612560164,0.2904961174767,2.760069706600243e-06,6.994314820487375e-08,0.6729352712811253,0.4091654409811372,0.03356191141218597,0.1094110817181438,0.007899826772762478,0.76656905581375,0.001417708759507125,0.01551859445215435,0.8848850579378277,0.01267224409612773,0.3708530072365235,0.2340222558961302,1.387153740825295e-05,0.3349244746871206,0.439547354223509,0.3266504341348465,0.242253856594513,0.0001282313360848829,0.003107797113722167,9.677251328444869e-07,0.2232168773360037,0.4710200755113078,7.210534548172745e-07,0.2120249358925813,0.4089371029704202,0.6785722661774016,0.01662462042946904,0.7590674946752155,0.1876537423161142,0.9241599531518112,0.06674332932227239,6.491777909485e-05,3.062956078833175e-05,0.1844908635008457,0.7427822978048343,0.3024646482496279,0.9335412831137162,0.001064095832737197,0.1165170090793727,0.008737653602988902,0.9667249776203444,0.2553394407389856,0.03068815178804911,0.003349224190106358,0.01456391478525574,0.005072467757866558,0.0004281792255491922,0.9034015663783298,0.08369300471674571,0.3867904718011927,3.6375637958394e-08,0.6529374910576278,0.9667249776203444,0.4216179731223939,0.01859796394536319,0.010811764752452,0.2257515370478488,0.8213858330296356,2.371231817572846e-05,0.1131777757594744,0.7532240459234625,5.478461642741834e-09,0.6699408684527994,0.1991228040821204,0.9682883608026949,0.3051662992780267,0.2235804904731974,0.003283854897631433,0.9308309839185359,0.6526129853827436,1.504924679095393e-08,0.5201175010310383,0.0003607185265296721,0.8243754036683677,7.819870330706447e-08,0.0001149606827412055,0.6874247431515486,0.03000554400539682,0.09254635138857564,0.01313968670295229,0.5248808959194099,3.373378800132064e-10,0.1515963583593026,0.001993233974349716,3.373378800132064e-10,0.06360742342570713,0.2252633600978533,0.6639015632948181,0.877604482057851,0.7535660221128655,0.8714900036831484,0.03602404001490216,0.8415730103256502,0.0002591712137223367,5.706589300652197e-06,0.01065264841665101,3.852137820223769e-05,0.3219650807577676,0.194007071969963,0.409193791359498,1.091639663567549e-05,0.9237745872021731,0.07211509036241204,0.6281951612456689,0.1085544587582328,5.635506427023269e-13,2.165570920820507e-07,0.01877171745488679,0.0006434700078248472,0.02268440163512287,0.7343471238202036,0.7946670231009676,0.08033377365844807,0.08972463673135223,0.6504456818213786,0.2037085667190542,0.1024056811554323,0.0399848012240607,0.4742184746149622,0.2004505626959373,0.3860102808802917,0.9774412545632509,0.003539989000976861,0.5199415460887471,0.1014363136006785,0.007573483835143147,0.02086832618696924,0.1120645953063172,0.08651829704377274,0.6764007425132146,0.08286691132058656,2.495679107195806e-05,0.07775280659276744,0.006487593724726159,0.4040321162379268,0.002970894902720263,0.0005207797323728224,0.008988709817766297,1.233786898892076e-10,1.571222768652214e-15,0.06739734342569061,0.2051350487443102,0.03564420662249792,0.8304129322816477,0.3173998670202456,0.4003591155312017,0.7149601501586884,0.01382415293859266,0.1085969712045524,0.0700569275904127,0.05357233377589301,0.7698107424344165,0.5386945957132041,0.01138641616213803,0.0001081476997320894,0.5833995778009018,0.2746862426537112,0.8849671031508964,0.6468875632467485,0.8249243555326579,0.543124666785875,0.1197898655709426,0.2504608827937322,0.09586001147213319,0.001430898289297457,0.09034839113254362,3.123352396308085e-07,1.609608055360358e-07,0.8571032135715024,0.9474643564167003,0.3762425906635552,0.003948747531562627,1.80162316812717e-05,0.7256152206180693,0.003302085642924687,0.7143098756699774,0.4931431299913641,0.6161898590496446,0.1763053635477349,0.1333807478841401,0.6640334098086184,0.6311911322870776,0.6394998111354099,0.3806907730961141,0.01578856228781265,0.1177108553463348,0.01130337176836267,0.155234476492854,0.8309135048809515,0.08124828509916732,4.521150292225786e-10,0.05957403212271355,0.6043248151185313,0.1799299572708256,3.458033939017872e-06,0.5602061003546376,0.0003058085176747016,0.4908757367707898,0.01349051080888683,0.06157630520470569,0.00220502099800771,0.195438006040356,0.04591250700966759,0.001059536009323774,0.004953023207190846,0.2202518991600624,0.02672665463644206,0.004354859747134125,0.6533111896164036,1.546760932733799e-06,0.2504608827937322,0.680310710399065,0.03876940174748266,0.07249918540759102,0.5121003998523723,0.5449417129155253,0.01215857551816579,1.419823103477099e-06,0.0001413929458043722,2.10524594325566e-09,0.121245769943816,0.6363733807376497,0.8989794641966314,0.01684667314013952,0.9475731803397907,0.08697830701094943,0.7524196837813943,0.9530488357227366,0.4880311325945096,2.571905032923068e-06,0.008881765128094072,0.001103111617383244,0.2767798952670429,0.1061731763566407,0.008728633231100734,0.006750839905327858,0.1113614189137009,0.4199169887323922,0.0005797508702575802,0.6212273509426681,9.284908033853821e-05,0.3434453052268933,0.958434954909163,0.3941236567628255,0.245015466704244,0.01839006815114813,0.2406353100505265,0.7118494704910838,0.1988806220638461,0.402192709540841,0.004566338417475198,0.2441367835960459,0.08859546280557644,0.4767717110575389,0.6255744040813462,0.2899356964264908,2.135066837489312e-06,0.4282247289513173,0.8930210967687493,0.9275369685350772,0.7828053743009487,0.4487151195924412,0.476756762596084,0.02447684327425029,0.002534949196414784,0.9506827324769835,0.1663373325102922,0.3401372035097909,0.01803401426875245,3.939259330507901e-06,0.359984743125948,0.4562599421848899,0.04342608748089905,0.01862796396220045,2.922054775997834e-05,9.065038146316224e-05,0.6389731408085969,0.00606777617489299,0.7056838401862913,0.02878588142437173,0.5246023052305091,0.0006352316860811252,0.002607440878573627,3.01070363270166e-05,2.459739185559778e-05,0.8005802367205138,0.5871524250479775,0.2726098729941036,0.9795889899768241,0.8184489812899766,0.5414620756962666,0.8257674793457406,0.307413319066177,0.3853730566115768,0.09653101364858606,0.5089555315046774,0.002476241544372525,0.2537536176506442,0.5397149447284727,0.9334972636748305,0.3650529857395265,3.365247188193935e-15,0.03346529502321031,0.5616118137834102,0.3822437158387152,0.008477416991579022,0.01075511973150209,0.8419292150864068,0.9402623611729388,0.0001093964244078748,0.01452931061547639,0.002154631946968098,0.000744415649473846,0.2682227382139072,0.0316512876645105,0.007031174375238802,0.01879134764037521,0.9413742407396022,0.02592271036449623,0.1502424321714859,0.523540688858436,0.05985385540134286,0.8531655109870756,0.06818026995287874,0.1129982630527059,0.007764343313259177,0.9834493731662909,0.6497213725458773,0.02645203496758951,0.6131549202660144,0.01927814235089696,0.22497442123457,0.06853355689801417,0.6855937234634061,0.1797697610722665,0.4072584594618684,0.3061764770169119,0.08210240418688952,0.008140994852431575,0.2626637581951933,0.04315099550487222,0.5967277656411298,0.09450240291908599,0.236703160320246,0.6345206647050011,0.9865358738424717,0.0003663992001149855,0.003756853530698788,0.9680025018692554,0.05054920761876507,0.002012794647176453,0.02113753060521363,0.3188216805619071,0.5250652348344655,0.1097575162918341,0.0005067147458134958,0.2726098729941036,0.3128622657331878,0.001311327113679115,0.0269643202659728,2.231238326187516e-14,8.855941755099222e-07,0.5735011626088767,0.001656869995112922,0.9203245028968917,0.02436442444673299,6.046867018934735e-11,0.02584815253703613,0.0002301701775994027,0.0006689152996905439,0.1060253362782586,0.4242863564631523,0.5530394613769887,0.7352698603352271,0.0008169222643398387,0.9512038837569853,0.8197058301033154,0.4654348305047873,0.2578748588907945,0.30379692084183,0.0004747135592545212,0.02126246524099589,0.3608126149729783,0.08269806661713487,0.01810462141820729,0.006900242125443201,0.2610751442039246,0.9377296281998976,0.04799519813833389,0.3255082891443709,0.03769689039782969,0.2704138224478135,0.01207944092679053,0.2202465227640047,0.2873025166759224,0.1891168931522433,0.4980729888585703,0.1433189239755163,0.5428267098070193,0.0278765488609047,0.01619546190626917,0.5521251202699506,0.0236458323638729,0.2474932975299636,0.0002139013783103786,0.001776483174872647,5.919951134992126e-05,0.3965397534891736,8.631075207199433e-07,6.009903968819766e-05,0.08512996392138643,0.6287885868447767,0.321191878949958,0.01238025102322004,3.946233129130797e-05,0.1177326739860021,0.2424743358000942,0.0006233270556811438,0.3610208578382436,0.8684518700709626,0.01227659316365955,0.3773395480590458,0.1223894163240823,0.232325613835414,0.3872357388573308,0.07025620239207593,0.922861263378296,0.1973415182140326,0.001637723398937906,0.1585691708811548,0.8849671031508964,0.4554854556477606,0.8230174961835058,0.005638814247062336,0.6529374910576278,0.9900376432841924,0.01129119466197781,0.5909563186830277,0.001128220784887271,0.1517713887751674,0.4517379352056182,0.1157881774774886,0.001417708759507125,0.8513414482102312,0.3912946725143422,0.7567571304934098,0.6847826684998352,0.01348412249621363,0.6179109156739868,0.6986542641793809,0.007622683478730377,0.02844878745723005,0.7907348947414258,0.3475223102317266,0.8407799456689329,0.009681246405027193,0.4043245167505359,0.04020784776544505,6.549958084490455e-10,0.3488614770677054,0.474954911976223,0.0003833828640564619,0.1506336284502748,0.4966945496989604,0.06046483284210526,0.6131549202660144,0.2775595699496495,0.1283650144110286,0.1174024717794584,0.9283305777772706,0.05229872882493589,0.1559173261612475,1.506722737375321e-05,0.3456556492902566,0.1952184369573112,9.890408699859639e-06,0.001188756786685886,0.1759644959050685,0.8580581648319041,0.2354825386390249,0.8995495712146644,0.3321171867485014,0.237016902345304,4.039474534468309e-11,0.4943561980797452,0.01648780940343677,0.0002692807283884061,0.07534438256582134,0.05192798259793301,0.2191697892178552,0.5499888130314559,0.8614577428626663,0.1223894163240823,0.745515803678569,0.8849993374797637,0.6299050465983622,0.159185763124334,0.0568249489110163,2.386610963550538e-06,0.1095323332716939,0.9531832963186443,0.489711039859958,0.8609507870379646,0.6629571868009061,0.8114813892024398,0.002896818553093327,0.1382020530737617,4.395239174839273e-08,0.0002010808989402309,0.002620599648890895,0.2225569449213017,0.5624278832261533,0.3163501129762324,0.08908573316357292,0.00116137641156875,0.03509843012537341,0.00935043273330481,1.363678969946935e-05,0.6987942635554578,6.084977737437176e-11,0.3166786180462445,0.1367586546945148,0.05345908272019278,0.01982512507101882,0.184664859884142,0.0127289177871424,0.491626281276503,0.0001778541301602575,0.9929442453063613,0.2971431013410861,0.8516649070122478,0.00295983607715052,0.01930806982638255,0.2439538687107402,0.6279536236026422,0.2207607351600009,0.7563463596578843,5.483898205385495e-05,0.2941231351606834,0.5170535852276568,0.1047382743885361,0.2303258829514211,0.03247962570785792,0.7203530045103602,0.009836092457967157,0.6311911322870776,0.5960392313497541,0.08435560401405175,0.1920607812991795,0.01307258518190895,0.891131492131317,3.276304243204267e-10,0.09650516904306981,6.434906773659445e-05,0.0004838966922792855,0.1291876176155664,0.4836684503654015,0.9490684023404511,0.2181597196824768,0.009459387135049173,0.06106726884899224,0.2980593561963169,0.001623255694028715,0.980324187467954,0.1663399650656264,0.1386083542201288,0.02857671304939728,0.736294258669271,1.804256368958376e-07,0.4318984007727337,1.549722129165928e-06,0.1175420283261645,0.210206255311333,0.04089506320823597,0.005025624349870587,0.3044752600607471,0.2636150752012038,0.05656823006202773,0.1989783603233743,0.7397093002232129,0.001759592660570554,0.9028178605484292,0.1297006164377255,0.8385973694115725,0.874664028679611,0.8923799038369623,0.1215488956225074,0.2571431895832408,7.844641585592257e-05,0.1004665110518029,0.04467901882820745,0.3807043441474092,0.06990406178051659,0.06846548994901847,2.528932401079638e-09,0.5439803559341565,0.116902587789953,0.2281037541609379,0.04046941330144945,0.4593410357649612,0.0103744052131774,0.5793737155578007,0.7281410384841436,0.7072353859187743,0.0004608364480513965,0.8671632096073962,0.1691210780255656,0.01829391706596991,0.02738910145884618,0.8843249868668023,0.7579522841309503,0.009465132766887763,0.07575346427819955,0.5090544110383687,0.3995968285332285,0.04905613155071811,0.6299246048065863,0.07267299614197019,0.3625016212859888,0.01368719354495265,0.7182121323420105,0.01323454356090736,0.1606480022348362,0.2048032327097582,0.020945610299228,0.09391527682282685,0.0005810557414218052,0.8260024114821645,0.3537654985620853,0.3641145303917463,0.4420509286511208,5.540253738412179e-20,0.2912526175518945,0.5701644717639186,0.185812240674818,0.800877068934901,0.004044431430970676,0.04693533831831481,0.09106998247019892,0.001458935335479238,0.03773658314892241,0.7898557883297671,1.167761125216789e-07,0.0002090942968535982,0.3294198849583681,0.3572563322230633,0.8635402840810823,0.4370089936682969,0.4026721898726306,0.6498399416086346,0.838281589565302,0.0002776567727803432,0.9611449046600512,6.347048328198691e-05,1.233002053715574e-05,0.2881254976984522,0.7293008845257715,0.526308112805806,0.0229677986343546,0.004972588564047166,0.4940663295053475,0.7318732816588687,5.480090014078873e-05,0.1057767839866205,0.3214469688876656,0.694945590484301,0.3904606910266455,0.5910193514889271,0.1899063003805614,6.4309284864715e-05,0.3005335836948509,0.08806078092851055,0.227892846607932,0.8845042095422153,0.853780991466744,0.569960469833415,0.005160519702512472,2.748334216108846e-07,0.005265217655995512,0.03050554352173116,0.8438001595498383,0.3967615292120792,0.0169378879703443,2.143551184580172e-06,0.01223920658110021,0.4596148143771715,0.0180837157476095,0.1092330319922355,0.002195832834098739,0.847256274340796,0.03792877581295546,0.7057438078834611,0.9140495083200286,0.5289988676810223,0.4070972001416223,0.7096108717234677,0.2050052568152988,0.4731452843170634,0.230646887173564,0.8630104100496064,0.5579336835022708,0.6508354136557793,0.3750566997993936,0.04993391676909768,0.8417671936750322,0.0008744085870950085,0.004863153631266094,0.2709790972746584,0.01155265375668624,0.7209511970999812,0.2746062141368557,0.03294148818846861,0.8370735154036125,0.02522931327763726,0.03316897802820055,6.107047319024079e-08,0.01957644575272414,0.6149434691727753,0.276052352348142,0.00580069883985365,0.3544523669225681,0.08119577358993281,0.9563938413518758,0.5205723731953527,0.01108308169707925,0.8714900036831484,0.4012108483660008,0.2983059596981751,0.09036125827570135,0.02904497746078482,0.0002171468140830478,0.006628323979929631,0.2875734196101148,9.554021943207153e-11,0.0001259822629279637,0.0002060379498941342,0.1013718710992879,0.009378026829767069,0.3517035730821587,0.8539762813970959,0.06185044465329658,0.5937828438807526,0.3628737505933717,0.1175058694217886,7.424017366609586e-05,2.61057479148272e-09,0.007529650526435228,0.000285434396683692,0.0008109979765750663,0.007500666419494252,0.335659953693635,0.09427304838261483,0.01274453427106579,0.9145377588448481,0.08422020471746484,0.1113641839932099,0.9680025018692554,0.002233849702348941,0.6355748218035313,0.8124816967345072,0.06827149325702625,0.118761966452148,0.2039021323449248,0.7073569545215106,0.3685467416677955,0.5810604801569503,0.004384534722658638,1.322559693221783e-07,0.9031345570029817,0.5048799719379026,0.01893040087293752,6.714752335591138e-07,9.398657249399954e-09,0.4072560509007686,6.165189335742932e-11,0.8903516629487274,0.2447286692703358,0.4280913236141939,0.06912362795252523,0.06992183925829623,0.09608622437614228,0.3900662005536362,0.002985890320717709,0.9596151131449897,0.3198482032246849,0.3081560869492858,0.008458511169051185,0.3874349415799883,0.8910399672511812,0.8152983654344619,9.960280672215792e-05,0.0006078258707302966,0.5336574075180006,0.006357375151616038,0.3706515435240225,0.1952184369573112,0.1313347935595998,0.1827465685909014,0.3720166279877956,0.6246357500878706,0.1549500703498756,0.3750566997993936,0.09017996990333098,0.2557051337154058,0.003900834746598609,0.1692749099649995,0.3005556899293212,0.1633212782436842,0.2226461252128384,0.00260584831385753,0.2522821225712149,0.4341811352126108,0.001317902713393793,0.09313735972188794,0.5753415958867029,0.3332533381388386,0.6421127216926329,0.05698336532269908,0.008297418174932618,0.003870828089844828,0.7287916591449547,0.1012365939305218,0.2643471155973277,0.004057909245011869,2.081790490518212e-14,0.4220067851084847,2.084807399241698e-07,0.04129718394952778,0.9197626857534269,0.2172723795338385,0.07097043872331162,0.3946826669058223,0.2440613007899664,0.9069463543390744,0.001621326796984096,0.6777390035352915,0.9444945158946495,5.675230070475227e-09,0.001393307236311265,0.0001248712531458483,0.000486402360271452,0.4365524786507405,0.02744510449655565,0.1068500034140608,0.8689473805076988,0.6473057297781399,0.02284426379753424,0.9399960630067151,0.1182502279764735,0.4395477240900566,0.2192722578888248,0.2382333972092383,0.8475762831286417,0.3292401402022583,0.384164518039348,0.4243107465585528,0.5435378539930549,6.508080249994613e-06,0.000308834766120304,0.9157018715813163,0.7140703435069609,0.003870828089844828,0.6697996345221027,0.06749932134609768,0.04451858365277369,0.5062905263380634,0.5129348731313284,0.5653220934179769,0.1994680024075363,0.03815504653187678,0.01978307419929105,0.01708100537267731,0.03801226280322961,0.4470519721316028,0.1206772679589798,0.0003320284231353615,0.01452228799011725,0.0001707433151811537,0.0003947509730270418,0.8176538182774842,0.459282079725866,0.5753415958867029,0.5266454985762038,0.3334040996146264,0.8185758309374994,0.7159651459700701,0.0001403405672479336,0.7787138678519878,0.3876459293074815,0.1266731838251142,0.2421778608224167,0.01817083486196575,0.05906164070416568,0.6464603278615149,0.00308199686727864,0.04356541801191764,0.005730051497529126,0.1479033020752943,0.05864390854219585,0.003741404599278658,0.9931593647709804,0.08092883573301332,0.276052352348142,0.1769471926373819,0.3196123185561531,0.0003731335523806806,0.1004607480816794,0.2021247568271315,0.0173827537416658,0.01167686101525353,0.06410722479369498,0.006249111330587902,0.001880534959445557,0.02878404116469898,0.9308309839185359,0.04480920031074873,0.5702861112009614,0.6865303866721716,0.161135359735264,0.002217244725184532,0.9724916132803773,0.03310058879374388,2.499405576462924e-05,0.8843249868668023,0.1448027116450414,2.098644321585647e-07,0.7637858610494423,0.00052031596641664,0.1386988155113719,1.734364957229184e-13,0.001031396794270317,5.952702638118534e-06,0.1816300481657555,0.03397284270558872,0.7927786657007155,0.0001473846161873151,0.7327799837658339,0.1748703849396694,0.07419512078504605,0.004257223224507516,0.08399255741019261,0.1186434283787427,0.0004841534805512125,0.06735735830984382,0.1270948583195243,1.608128904541093e-07,0.01721355606497938,0.03201890900449889,0.1825112615828148,0.6136620450636623,1.495612557282932e-09,0.02357874924869052,0.009236679650141157,0.1808310898366843,0.2028370952229857,0.6699408684527994,0.1399741834231116,0.1004334210219624,0.1672570040156478,0.9844489058736869,0.005516913438258793,0.3211687514007098,0.05275508299402507,0.00488275779468696,4.387634046612761e-05,0.002746910209481309,0.1261251500692631,0.05924648455655371,0.5726407337823777,0.27928842764904,0.002272738850679136,0.1705939240198945,0.06050286485029305,0.6566545328064539,0.5255331836314646,0.5510656472186992,0.005829897841558014,0.465597477607034,0.844400565870377,0.1787091154939428,0.1081438487060412,0.00482272629515692,0.2014424711501141,9.784835645256384e-10,0.02299493849493858,0.05422444800412433,0.0536800076603319,0.8843249868668023,0.2168784753567427,0.8506495279851128,0.8535657381616963,0.1351249712988475,0.3441470098833407,0.3586085363890772,0.9623772842329845,0.9695891793069894,0.6928056191072733,0.0003526914105954796,0.0002123404317665458,0.7737001754775076,0.2911616945652905,0.002103129207788701,1.552894624464422e-05,0.07801848743555102,0.07929702170106022,0.565894056900074,0.07470778452157176,0.2090384253676418,0.0008627061704859335,0.004364737463330549,0.06158672336795018,0.4913420629736113,0.07422382678413932,0.03307211848550726,0.005011668826860729,0.9234085106683,0.001301274912046833,0.1746292140487166,0.08475378509618454,0.5373609453489389,0.6980315376605691,0.3360269504515609,0.8947613202575595,0.002716085175238089,1.418822151962079e-06,0.7087266231513016,0.001445735740554856,0.9028178605484292,0.03875302972824465,0.1108058180597372,0.1277444609048294,0.0421708321575214,0.3694354657866683,0.7742147265460743,0.02622422553912665,0.000216819736417688,0.001417708759507125,0.4386289441953353,0.1096871489572996,0.09249757476310387,0.01644196026024882,0.6528295936850514,0.150317841809961,0.01948094265758028,0.8910399672511812,0.01862463754274417,0.001844870130544599,0.2693889002855231,0.01310604817454241,0.9413742407396022,0.653710137159073,0.2690987818775205,0.2282269055351131,0.8048763544433283,0.3967753224876497,0.9474643564167003,0.01115949087821304,0.01198104690033338,0.01401640762087456,0.1764043048698077,0.8468209845008692,7.939972912436976e-05,0.04188432262760428,0.01578096629295968,0.6254832416658821,0.9873634005133405,0.02640458443545643,0.1117463329133245,0.02689318719701441,0.005599893480274032,0.9241599531518112,0.7834062841595261,0.008288065635184097,0.1648424912747919,0.06387513538968383,0.6734434660698077,0.8513414482102312,0.9457103245432588,0.002942519690385277,0.0001032191888188289,0.001225573696562392,0.001218782933205942,0.05651924095832213,0.3629937620115812,0.2636150752012038,0.003924787247901241,0.2886657176630782,0.2599782784623171,0.09235755768270505,0.1667820590189552,0.9449580636486389,0.002423703240614098,0.07562572563208092,0.2726098729941036,0.05035544625469826,0.03088942672422952,0.2411408376233609,0.1083042441465824,0.70558544842823,0.5246023052305091,0.4970982709230296,0.8023517781456845,0.01306730484842443,0.01815204505309521,0.8653034871842542,0.6696158642936901,0.05030338677749385,0.0002356680883189522,0.07140706044199885,0.9965983444101834,1.528625306054041e-05,0.9854870976918468,0.0003283318665064851,0.5291811815894597,0.6620711622399017,0.002796447405321597,0.5037205149999638,0.1645065101723163,0.1125940988341855,0.9282757403679216,0.1847720355685589,0.02342837554297137,0.9335485948274168,0.001539236712265653,0.2354105842321524,0.008244890827869141,0.2770858857842211,0.6974020472628388,0.0003707563824047295,0.1676474638617765,0.2636150752012038,0.0004478600999683508,0.5401958296979444,1.547675458508298e-06,0.001304227970322589,0.6612962027353875,0.03323549159929477,0.0489294400673741,0.01959805373153822,0.2752043016451501,0.6590773652780622,0.9308785993589326,0.1611976029390919,0.6856895868122939,0.06329677784707065,0.05951505012945902,0.7590674946752155,0.0008496592116564966,0.3167243990651296,0.9226981120463438,0.574743576204624,0.8007572903032278,0.5013018860753161,0.2613689302714907,0.1920604662704651,0.849857274950241,0.4219998600057828,9.346580097323073e-09,0.001406550041491391,0.5523917611279887,0.005187950576795181,0.03283213251033416,0.6785722661774016,0.7408635786055862,0.3658640204611395,0.1068420461172508,0.4537621409470914,0.1150033270325992,0.0007305101472009756,0.342382340775663,0.001691938422113464,0.001983917635677204,0.5869931255293354,0.5360346082223635,0.9238851059101465,0.4608988228201583,0.1458285857048916,0.4528564738871182,0.8897905719035331,0.4501269560216393,0.9350595769451101,0.05193225374791457,0.1388538499480837,0.1207370828100211,0.8876333576935422,0.9193779782719373,0.967160322054779,0.4368443455856744,0.1935196340847538,0.3673403597761943,0.2815861144021491,0.0780169817122511,0.8526612841289106,0.9996962035915474,0.09002098889698716,3.864743176618661e-06,0.4208810612254125,2.157501252986107e-08,0.4942091505636673,0.847256274340796,0.005459302238844019,0.8706224815918167,0.2643456957423613,0.8811348616346973,0.1635721756001148,2.673977689417029e-06,0.7139366268741977,0.7276153334763976,0.01694836511625197,0.2608271952383056,0.02831257498635087,0.8519509027606581,0.5793737155578007,0.224468315644199,0.00414027701995673,0.2998490742934766,0.3661277489949646,0.06214369428147353,0.9028178605484292,0.6421127216926329,0.02356535450158217,0.1466790974168839,0.1236556265086363,0.7383365371553285,0.02363823099643695,0.2432717669613699,0.8071879435803547,0.01038482144958714,0.9821447228025674,0.4616806481753231,0.974575736568667,0.6240630752870969,0.6343336173846293,0.2595517523105654,0.3771910216276448,0.001201811169104761,0.972144603652311,0.685339599623771,0.007096438114728727,0.003605291420464515,0.2632082190657489,0.1287898599361647,0.2042183304769264,0.03496285167939311,0.03014398577637032,0.08697830701094943,0.4155943084013178,0.9004135330176521,0.3611067928513189,0.4217085000328353,0.4159765408270434,0.08342891627591603,0.0002897790373084506,0.1377441080788345,0.1156790803681256,0.05839382182058429,0.20342189182461,0.0008406865074105851,0.6629571868009061,0.1455334102451411,0.2878734064353504,0.06454599267412504,0.1779877031020922,0.1868574268258571,0.2317388546417266,0.8348274033402979,0.6832753989278405,0.02298108685538162,0.249504056560023,0.9908278124918487,0.1052030898985916,0.1602958903110953,0.2122030512462202,0.943397327718118,6.81963133198243e-06,0.1985215985090942,0.00569084459921002,0.3405915008958249,0.5441267241231195,0.591092588756965,0.903486632302863,0.06730714611629765,0.2821566895196074,0.7745327958319056,0.9453277826064788,0.726829551498742,0.9894953227737648,0.09330606292358848,0.0002619781375480203,0.5058171370863256,0.01324640000907825,0.005776204376819817,0.1112398352690021,0.001498587806377068,0.01199289156651527,0.01089176459299387,0.04490607361517637,0.6869528381966384,0.3693738852823885,0.2881375377673629,0.04083275356718834,0.223625547441959,0.03480261476320932,0.3634255908321015,0.01088019081219225,0.615676226177041,0.04295230065049436,0.4260516005251273,0.003576269053717969,0.002772456340056832,0.537745600961298,0.0020135154816408,0.1381015030246562,0.0008415405087383476,0.006394137532104063,0.2377185983838752,0.2302309868104189,0.003707512796916751,0.1435350739989242,0.4506703524668473,0.1266640065396992,0.943397327718118,0.02250037740049012,0.1969446021636529,3.354782051932218e-09,0.1722007780569176,0.06283201509638636,0.3456495380928407,0.02275411295744748,0.9197626857534269,0.2412267984006883,0.9757496253942517,0.8964506382430526,0.5254522968736417,0.7601932471678808,0.4318810840625703,0.01578041042190493,0.1627164806640051,0.3965171524233889,0.339959901992968,0.3685467416677955,0.2312749923693553,0.003907595291854639,0.2269314656210225,1.197486954082552e-06,0.1142464982387263,0.001089968831106073,0.1485291077260198,0.02584755766550295,0.4876646992206556,0.106401380450318,0.01076217349688606,0.01499383336619971,0.2275193794844247,0.6734434660698077,0.3140809319625932,0.5298873798088661,0.3564722793637772,2.293191971586901e-08,0.2900329707592649,0.1252024565065581,0.3114829011715899,0.4069610973209792,0.1228404118544715,0.7023725341534056,0.006665265473957617,0.3551479156464431,0.01401362515374122,0.2619636522715194,0.06202787324309633,0.002181356164071559,0.5818929073062236,0.9755278428826513,0.9117619935249506,0.2798314709486607,6.156516630902866e-11,0.8497249732880033,0.6384920226250854,0.5195021639477211,0.724627761971077,0.3511062444832618,0.005449603729898886,0.4001557657242756,0.0277903836203813,0.5431832419984362,0.8886025215690979,0.9880253661309057,0.837678263996971,0.07493845768001835,0.8814955251191607,0.8226723760557845,0.0003718667918918406,0.5021980235554665,0.004881610334806999,0.8371178248661907,0.05229872882493589,0.03064029892808758,0.04032725523079084,0.00788686479997565,0.2979196277382847,0.3497590187301193,3.750411535333337e-07,0.8763313836932529,0.04385237700751179,0.1758096740171421,0.06014062586630304,0.9546679741895375,0.1955857059209185,0.2504608827937322,0.4031213562311736,0.7656189501447032,4.968855601295516e-08,0.1512745071108363,0.03577266395027816,3.208427779337512e-07,0.527114574205694,0.09841508087987126,0.6556158867943204,3.165417149602019e-07,0.009319120358153231,0.07120995151608908,0.4142440935275727,0.4463578166378016,0.241203257129888,0.9852364485023742,0.5300285111235087,0.01226527247538659,0.002425594569367514,0.5843618054141689,0.2855812680750078,0.003750700770603787,0.008316460197520413,0.02490228188073203,0.9643945384412765,0.1761523525887129,0.5009302398388578,0.08517450298089026,0.00260015862431067,0.2661657017572677,0.2597834095926548,8.654078917802602e-07,0.08038773607896682,3.169476273393437e-05,0.2322460106927781,0.0006728994896126104,0.389709378443426,0.0001811345833011774,0.7345404147880621,0.5478649921609348,4.361560562471977e-12,0.01383810067739076,0.187368749174264,0.6788264507371096,0.5546732744918126,0.042155983401264,0.0003325541419876717,0.5559038210648969,0.0009716201690033004,0.1902462343792798,0.9078770692133724,0.2418392527982449,0.7321776191578673,0.8292056809229637,1.204846053081981e-06,0.1897517466487762,0.4994976118551448,0.2097053213369122,0.1663481954551695,0.001566737687459464,0.3930667192227297,0.1593420083822676,0.007609475703629743,0.03652202214363206,6.162368216126103e-08,0.005904535155213403,0.6408172828474565,0.02064076782740632,0.4826164402784239,0.03867740963207311,0.6428387513857662,0.02197354546436048,2.457269203410428e-10,0.3442267592381981,0.1250653146165666,0.009603716658458331,0.462406887227786,0.411703827508285,0.8227056143931116,0.09583710730906042,0.6355105617908886,0.02142317171104394,0.005780697754675259,0.04294773671593684,0.0001375979655726341,0.771099084627607,0.005829897841558014,0.01255460922524552,0.2518305240640403,0.9211599188226122,0.3530536568724548,0.009308205306890124,0.6531957976286137,0.2769622755684615,0.001637723398937906,2.552310874006704e-05,0.7261114955102597,1.286185354506669e-06,2.420523410957966e-05,0.3440542314367259,1.033365096177553e-08,0.152897810036233,0.1194077324275913,0.9457103245432588,0.005459302238844019,0.3368653898630334,0.249504056560023,9.529380239256639e-06,0.2441367835960459,2.116093989307315e-05,0.02311062618860435,0.8129890457734664,0.8417671936750322,2.009973759195511e-05,4.541403214362366e-05,0.1731819733613915,0.1186434283787427,0.2854434214358612,0.2723225404574259,0.5428185704401048,0.7898557883297671,0.4037019766552952,0.7875361089515666,0.0003637248440422671,0.02589661185521407,0.02171825858432675,0.05302009812947486,0.7815576867706236,1.203533936401474e-06,0.01660600291004828,0.05175034835827542,0.06467939971855274,0.06733892996308277,2.481772410968349e-10,1.288420971678799e-06,0.004916448653352909,0.932462237550484,1.204846053081981e-06,0.7715267148079097,6.981951926974889e-05,0.8226723760557845,0.5843939254280103,0.5722814853840549,0.4123639719407735,0.6552587616820121,0.5570856569909384,0.2671953346533345,0.792537497716813,0.2276680868268261,0.1768434974115294,0.001087890354844499,0.0004971969547255175,0.6525106319864578,0.00063738238426599,0.9553308424446226,0.2185852004370389,0.0004177404994757411,0.7704808443925321,0.7071877197593545,0.1228022550050988,0.7532240459234625,0.9184690683936957,0.020481204863577,0.578973261511991,2.398501155559798e-05,0.0005029131869721012,0.0003186938063486062,0.2226445780796723,0.9028178605484292,0.9117619935249506,0.317042285378651,0.000953155086623929,0.8763313836932529,0.6088004503264968,0.109054095252291,0.5757249829426651,0.7801092185523277,0.132730336552082,0.7342618731014088,0.8481614257839557,0.4638532048652745,0.140260290367863,0.2226445780796723,0.6650667860967825,0.0183516726767906,0.09058404622080179,0.05268044449105929,0.4386289441953353,0.7143778801135354,0.02193070250749247,0.3539405064452272,0.0003320284231353615,0.08503004056970764,0.2284028070126589,0.6498399416086346,0.005776086105730303,0.8723868380994764,0.4046234114889801,0.01836604326910423,0.6450834411282065,0.1503611502819016,0.3977558966344911,0.05874782935501673,0.2007007748565381,0.09932631380663805,0.02299493849493858,0.8683234427644566,0.7522453855069544,0.9165336107968974,0.6944356402962001,0.8078670908756849,0.8197267833818732,0.8746744873767062,0.02671297727372138,0.3370547390236703,0.9577315007099367,0.1366350560992325,0.3003594773365184,0.9441497348709813,0.0003713283790853399,0.5147544552824809,0.5975514474254542,0.03001630516474221,0.1467987391190681,1.364427735721428e-06,0.3796961502662468,0.0001599276384413185,0.6401015791048864,0.7609494344372136,0.4924555908875572,0.8226723760557845,0.6919678220004876,0.9020177389532693,0.000370349016366549,0.2292160063115338,0.363069179274192,0.8448825728047383,0.8500844858652026,0.6311911322870776,0.9398932889846108,0.2647671271064567,0.2195510627334242,0.07822860695210031,0.6382332607027866,1.47402339133391e-05,0.06912362795252523,0.03829433144033625,0.5351924402671527,0.0001284789979040784,0.08498971473126266,0.6270576945222986,0.001299504029697177,0.9764593904835445,0.8940376292345515,0.1914797940354373,0.9475938747422845,0.6912671601005242,0.4081049363370035,0.1369183685000811,0.622381035708649,0.6887720950644894,0.519132942494467,0.2261051427775176,0.8589355940303313,0.1514077082328475,0.245440222764704,0.2115111233166761,0.3749364878892887,0.8418465431043597,0.4478461990219065,0.1964831858761206,0.1439712433742693,0.2611102414709787,0.5529396979120712,0.1986421645848077,0.0004068153545563113,0.4102980624461148,0.5336870471899419,0.1014818793691013,0.5569780421042256,0.9702849155720955,0.4455226039416476,0.2170009984121338,0.3401910073007361,0.3741974732660801,0.8843249868668023,0.1254659122994257,0.2943056285489974,0.1062169242110432,0.3851417537873312,0.9553308424446226,4.394070468872702e-05,0.8049355779302654,0.3494128855452001,0.5569780421042256,0.1399741834231116,0.8319226389368456,0.2587313558048368,0.1091956174004123,0.8438001595498383,0.005307586177546813,0.3384137302380361,0.4184237357692526,0.2382333972092383,0.01378307373095495,0.09378977987545399,0.7820993290376268,0.5149685355752622,0.3412776519222481,0.8466977292386075,0.1863288574043403,0.8981768256616506,0.6514098912560204,0.5382625463045708,0.06657382881187576,0.9687564876222832,0.7769166684898277,0.3553714626386735,6.116212148005591e-06,0.6160675372131708,0.007917853787377834,0.08517450298089026,0.1077353980695581,0.4087082349669418,0.01524679619072917,9.918376252053845e-05,0.6542695094187587,7.599554792586779e-06,0.5745221311696647,0.650999192346194,0.2330391872773991,0.0006901374753241941,0.219668422448263,0.6351943460337925,0.9261881631239571]],"container":"<table class=\"display\">\n <thead>\n <tr>\n <th> <\/th>\n <th>SYMBOL<\/th>\n <th>GENENAME<\/th>\n <th>log2FoldChange<\/th>\n <th>padj<\/th>\n <\/tr>\n <\/thead>\n<\/table>","options":{"columnDefs":[{"targets":3,"render":"function(data, type, row, meta) {\n return type !== 'display' ? data : DTWidget.formatSignif(data, 2, 3, \",\", \".\", null);\n }"},{"targets":4,"render":"function(data, type, row, meta) {\n return type !== 'display' ? data : DTWidget.formatSignif(data, 2, 3, \",\", \".\", null);\n }"},{"className":"dt-right","targets":[3,4]},{"orderable":false,"targets":0}],"order":[],"autoWidth":false,"orderClasses":false}},"evals":["options.columnDefs.0.render","options.columnDefs.1.render"],"jsHooks":[]}</script> --- class:empty-slide,myinverse background-image:url(images/desert2.jpg) .mytop[ The number of grains of sand in this picture accurately represents the number of approaches for functional analysis of gene expression. ] --- ## Plan for today * Gene set enrichment analysis * 1st generation: hypergeometric test, Fisher's exact test * 2nd generation: other algorithms * 3rd generation: pathway topology analysis --- ## Gene ontology database✪ .pull-left[ GO – Gene Ontology * Genes are assigned to GO categories * GO categories are assigned to each other * GO can be represented as a *directed acyclic graph* ] .pull-right[ 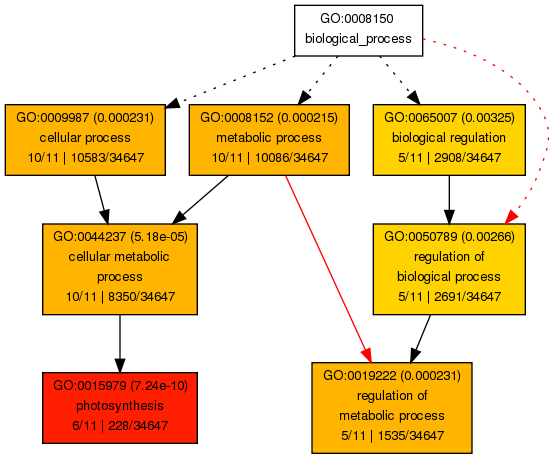 ] --- ## Gene ontology database✪ * Three main categories: biological process (BP), molecular function (MF), cellular component (CC) * Each category is a so-called "directed acyclic graph" (DAG) * Each gene can be assigned to multiple categories * Each category can have multiple "parents" and "children" (so it is not a tree!) --- ## KEGG pathways✪ .pull-left[ KEGG – Kyoto Encyclopaedia of Genes and Genomes KEGG pathways - a manually curated collection of ~ 300 pathways ] .pull-right[  Minoru Kanehisa (金久 實), the creator of KEGG ] --- class:empty-slide,myinverse background-image:url(images/hsa04110.gse16873.png) --- ## Other gene sets Different lists of genes can be obtained from various sources. * Coexpression data sets: groups of genes that have correlated expression profiles -- * Results from another differential gene expression analysis experiment: groups of genes significant in a comparison -- * Targets of a particular miRNA determined computationally --- ## MSigDB – a meta database of gene sets✪ .pull-left[  ] --- ## Group of genes, and then what?✪ First, define DEGs: differentially expressed genes (e.g. `abs(log2FoldChange) > 1`, `adj_p_val < 0.001`) Second, define which genes are in a given gene set (GS), and which are not. .pull-left[ **No significant enrichment:** | |DEGs |non-DEGs|Fraction| |----|-----|--------|--------| |in GS|5 | 95| 5%| |non in GS|50 |1000|5%| |Fraction|9%|9.5%|| ] -- .pull-right[ **Significant enrichment:** | |DEGs |non-DEGs|Fraction| |----|-----|--------|--------| |in GS|50 | 60| 45%| |non in GS|70 |1000|7%| |Fraction|42%|6%|| ] How can we test it? --- .pull-left[ **No significant enrichment:** | |DEGs |non-DEGs|Fraction| |----|-----|--------|--------| |in GS|5 | 95| 5%| |non in GS|50 |1000|5%| |Fraction|9%|9.5%|| ] -- .pull-right[ **Significant enrichment:** | |DEGs |non-DEGs|Fraction| |----|-----|--------|--------| |in GS|50 | 60| 45%| |non in GS|70 |1000|7%| |Fraction|42%|6%|| ] -- .pull-left[ Results of `\(\chi^2\)` test: Left: <pre class="r-output"><code>## ## Pearson's Chi-squared test with Yates' continuity correction ## ## data: matrix(c(5, 95, 50, 1000), byrow = TRUE, nrow = 2) ## X-squared = 2.0686e-30, df = 1, p-value = 1 </code></pre> Right: <pre class="r-output"><code>## ## Pearson's Chi-squared test with Yates' continuity correction ## ## data: matrix(c(50, 60, 70, 1000), byrow = TRUE, nrow = 2) ## X-squared = 161.1, df = 1, p-value < 2.2e-16 </code></pre> ] --- ## Disadvantages of the generation 1 approach * You have to define DEGs... but that depends on the number of samples * the larger the number of samples, the higher statistical power * the higher statistical power, the more DEGs you can detect * therefore, the results depend on an arbitrary threshold * What to do if you have no DEGs at all? --- ## Generation 2 approaches – "Functional Class Sorting" * do not depend on arbitrary thresholds --- ## GSEA algorithm✪ 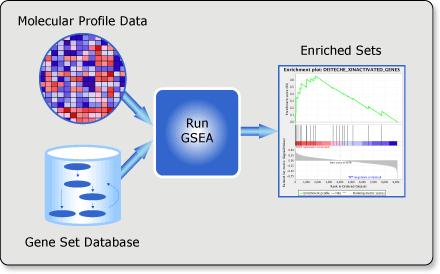 --- ## GSEA algorithm * Calculate the (pseudo) Kolmogorov-Smirnov statistic for each gene set * The statistic has a value, but no associated p-value * Test the statistic and obtain a p-value by a permutation test --- ## Permutation tests in statistics * Normally, for a given statistic (like the t-test statistic or chi-squared statistic), you can calculate a p-value from the known distribution of the statistic * If you don't know the distribution, you can simulate it by permuting the data * Advantages: no assumptions about the distribution of the statistic * Disadvantages: computationally expensive, and more importantly: you need a sufficient number of samples --- ## tmod algorithm * Arrange the genes according to p-value or another similar metrics * Test whether genes from a given gene set are denser on the beginning of the list --- class:empty-slide,myinverse background-image:url(images/img21.png) --- class:empty-slide,myinverse background-image:url(images/img22.png) --- class:empty-slide,myinverse background-image:url(images/img23.png) --- class:empty-slide,myinverse background-image:url(images/img24.png) --- class:empty-slide,myinverse background-image:url(images/img25.png) --- class:empty-slide,myinverse background-image:url(images/img26.png) --- class:empty-slide,myinverse background-image:url(images/img27.png) --- class:empty-slide,myinverse background-image:url(images/img28.png) --- ### Tmod statistics Since normalized ranking for a gene `\(i\)` `\(r'_{i}=\frac{r_i}{N} \sim U(0,1)\)` -- then `\(-\ln{r'_{i}} \sim Exp(1)\)` -- and `\(-2\cdot\ln{r'_i} \sim \chi^2_2\)` -- thus, if `\(G\)` is a set of genes, `\(-2\cdot\sum_{i \in G}\ln{r'_i} \sim \sum_{i \in G}\chi^2_2 \sim \chi^2_{2\cdot n(G)}\)` This gives a fast and reliable statistics with a known distribution which can be used to associate a p-value to any set of genes. --- 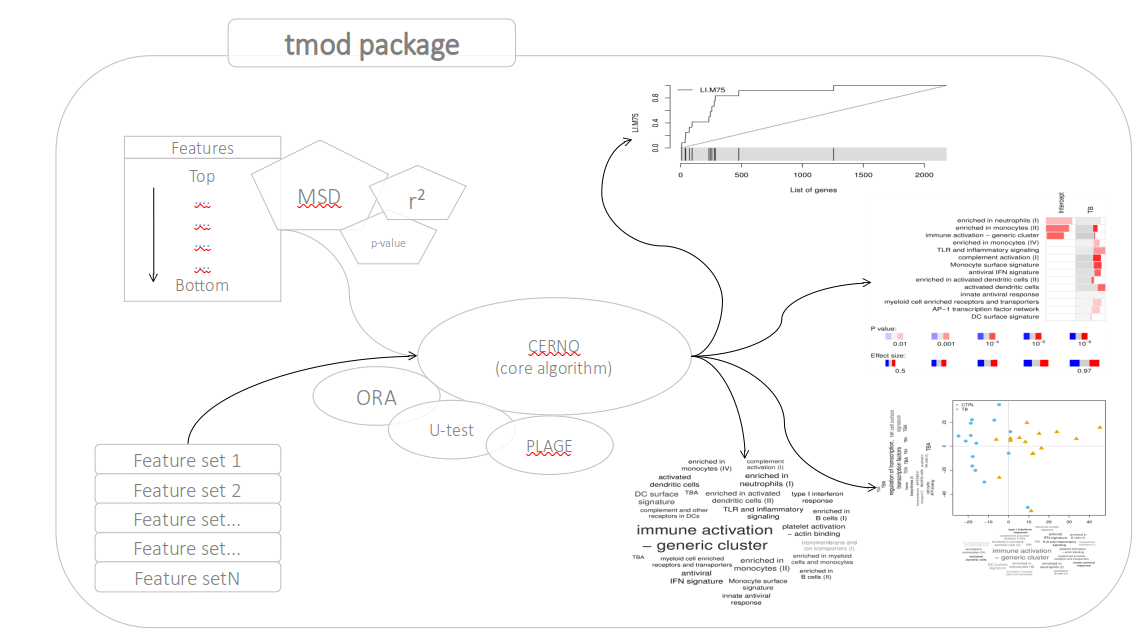 .footnote[*Zyla J, Marczyk M, Domaszewska T, Kaufmann SH, Polanska J, Weiner 3rd J. Gene set enrichment for reproducible science: comparison of CERNO and eight other algorithms. Bioinformatics. 2019 Dec 15;35(24):5146-54.*] --- 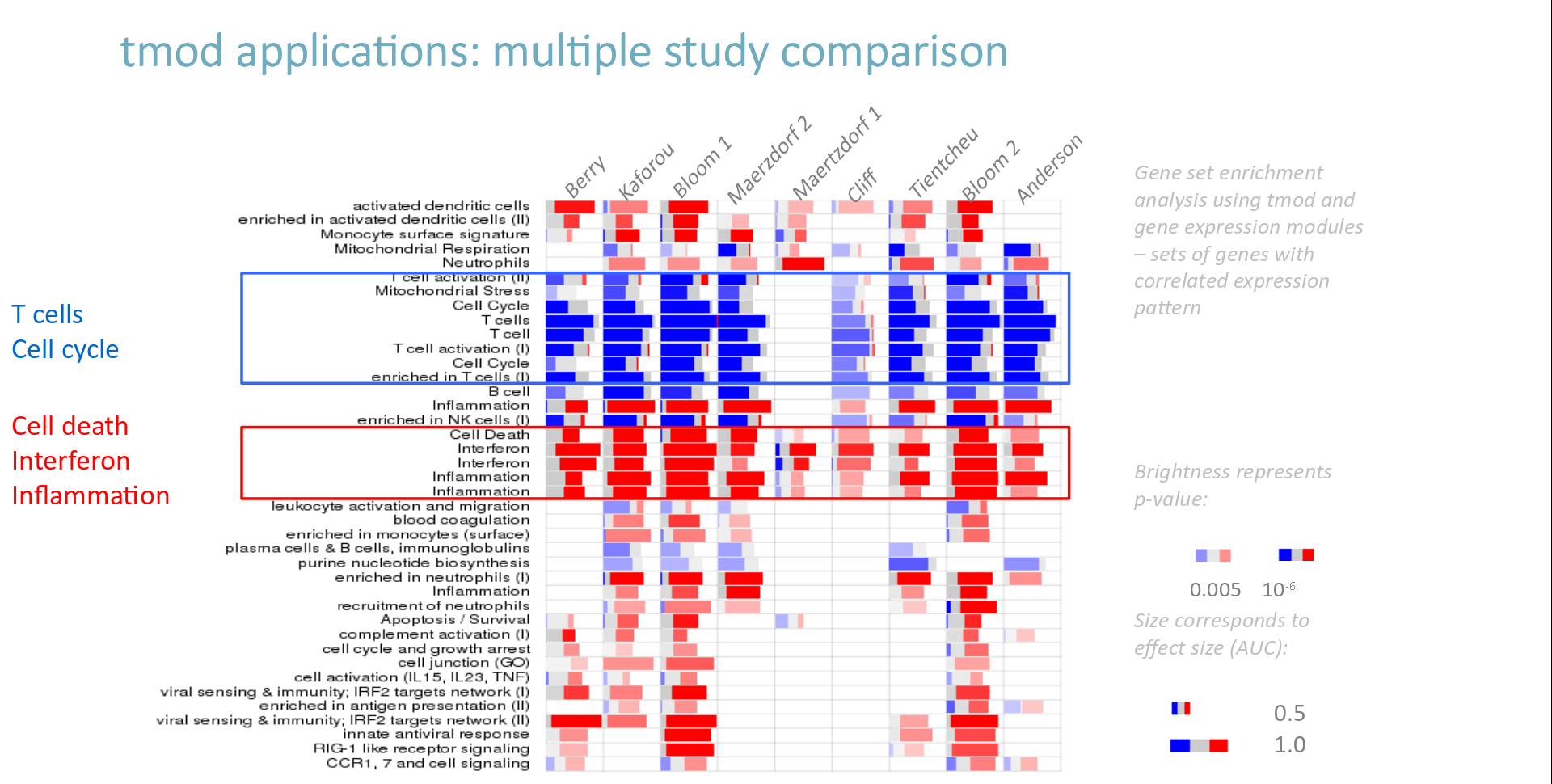 --- 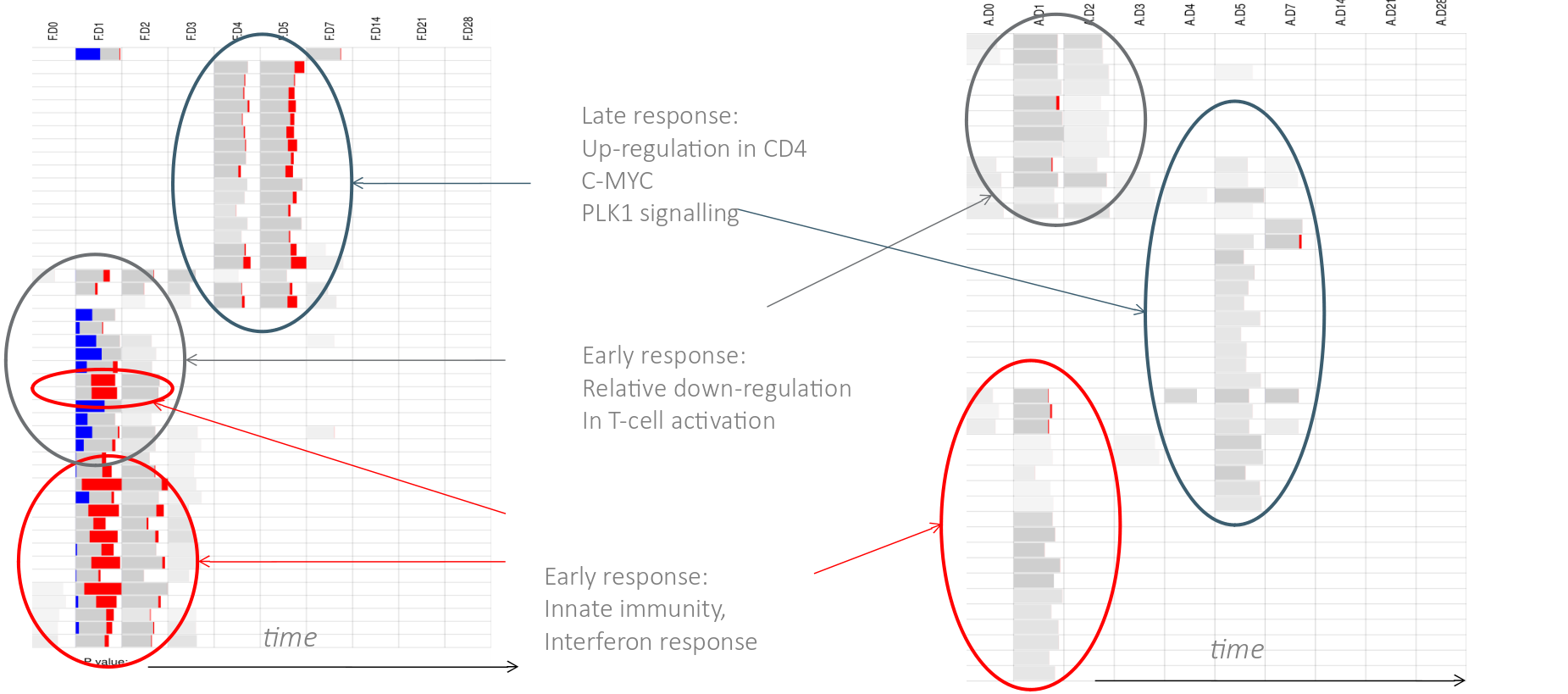 .pull-left[Vaccine with an adjuvant] .pull-right[Vaccine without an adjuvant] .footnote[*Weiner, January, et al. "Characterization of potential biomarkers of reactogenicity of licensed antiviral vaccines: randomized controlled clinical trials conducted by the BIOVACSAFE consortium." Scientific reports 9.1 (2019): 1-14.*] --- ## Network analysis (generation 3) Genes / proteins may be linked by * co-expression * common pathways * literature (co-citation) * synteny (co-occurence in genomes) * common motifs --- ## STRING